February 2022
![]()
![]()
© Crown copyright 2022
This publication is licensed under the terms of the Open Government Licence v3.0 except where otherwise stated. To view this licence, visit nationalarchives.gov.uk/doc/open-government-licence/version/3
Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned.
This publication is available at www.infectedbloodinquiry.org.uk
Any enquiries regarding this publication should be sent to us [email protected]
02/22
Printed on paper containing 75% recycled fibre content minimum
Printed in the UK by the APS Group
Plasma manufacturing processes, pathogen inactivation/reduction treatments, plasma-derived medicinal products, recombinant products, and therapies with these medicines
i. Example of obtaining a final product: preparation of albumin
i. Early method, with regards to hepatitis
ii. Results of a safety study published in 2011
i. Clotting factor concentrates in the 1960s – 1980s
ii. Intramuscular and intravenous immunoglobulin in general
iii. Intramuscular immunoglobulin (IMIG) and its indications
iv. Intravenous immunoglobulin (IVIg) and increased demand
i. First-generation immunoglobulin
ii. Second-generation immunoglobulin
iii. Use of chromatography in immunoglobulin manufacture
iv. Intravenous immune globulin and thromboembolic adverse events
v. Are all commercial IVIg products equivalent?
i. Virus transmission by plasma products
ii. Clotting factor concentrates and virus transmission
iii. Polyvalent immunoglobulin and virus transmission
iv. Anti-D immunoglobulin and virus transmission
v. Variant Creutzfeldt-Jakob disease transmission via blood and PDMPs and risk assessment
i. Frozen and freeze-dried cryoprecipitate
ii. Factor VIII and Factor IX concentrates in the 1970s, 1980s and 1990s
i. Structure and function of Factor VIII and Factor IX
ii. Factor VIII immunogenicity
iii. May some Factor VIII concentrates induce more inhibitors in patients?
i. Alloantibodies or Inhibitors
ii. Modulation of the immune system
iii. Anaphylactic reactions and inhibitors
This report has been written by two experts appointed by Sir Brian Langstaff in 2020 on behalf of the Infected Blood Inquiry (“the Inquiry”). In the Letter of Instruction from the Inquiry, the two experts were asked to respond to a series of general and specific questions grouped into sections. This report addresses the questions posed in paragraphs 9 to 12 of the Letter of Instruction.
The report addresses the processes of fractionation, fractionated blood products, heat treatment, plasma-derived medicinal products, recombinant products, therapies with plasma-derived medicinal products, and a number of other related topics.
The experts are aware that biotechnological production processes are complex and that it is necessary to use specific technical terms in answering the questions. Efforts have been made to reduce the use of highly technical terms as much as possible and a glossary has been included to assist the reader.
The questions in the Fractionation Letter of Instruction are provided below. The full Letter of Instruction is available on the Inquiry website here.
The following section provides a short overview of fractionation. It describes the composition of plasma, the proteins it contains and how they are used in plasma-derived medicinal products. It also outlines processes relating to the two different sources of plasma: either recovered from the processing of whole blood into components, or through plasmapheresis whereby plasma is removed from a blood donation using an automated procedure, with the remaining material (red cells and platelets) returned to the donor. The final part outlines the European testing regime for human plasma.
Plasma fractionation is the process used for industrial production of a unique set of proteins of high therapeutic value made from human plasma. The clinical uses of these proteins, known as plasma-derived medicinal products (PDMPs), continue to grow.
Today, fractionation includes all the steps involved in PDMP production from plasma, whether or not ethanol fractionation is the first step, including further purification steps and robust virus inactivation/virus elimination steps.
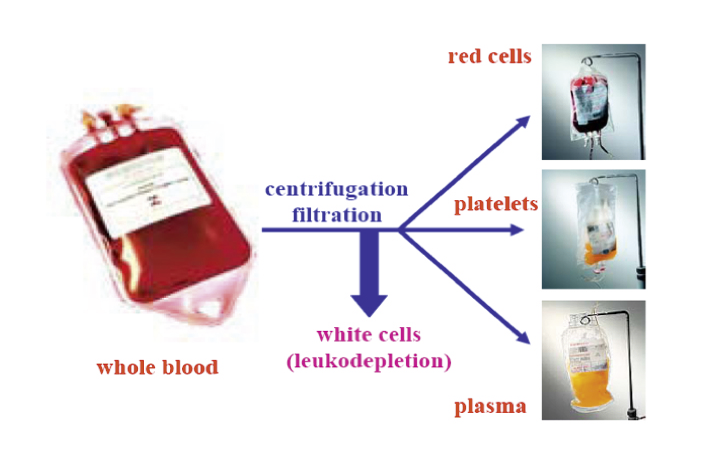
The whole blood is separated by centrifugation/filtration. The resulting components are 1) the upper phase, a clear yellow solution (plasma) at the bottom in the figure, 2) a thin layer (buffy coat) containing the platelets, after leukodepletion (removal of white cells), 3) erythrocytes (red blood cells). In the plasmapheresis process, the cells and platelets are returned to the donor.
Blood and plasma are collected from donors at blood or plasma collection centres. The blood bag for collecting whole blood contains an anticoagulant to prevent clotting. After donation, the plasma (the liquid part of blood) is separated from the other blood constituents such as erythrocytes (red blood cells), platelets and leucocytes (white blood cells) by centrifugation (Figure 1). Human plasma and its fractions and derivatives are used in biomedicine, as plasma contains thousands of proteins. These constitute the largest and most diverse set of proteins that the human species can produce, performing a wide range of functions of which some are still unknown. The absence, deficiency, or dysfunction of a particular plasma protein can impair the homeostasis of the human body and be life threatening or disabling.
In addition to the “classic plasma proteins” such as albumin, clotting factors, immunoglobulin and fibrinogen, plasma contains all tissue proteins (leakage markers), glycoforms, proteins in their active and modified forms, and those destined to be removed from the bloodstream.1 In other words, it contains the entire set of proteins that are or can be expressed from the human genome at a given time. This set of proteins is defined as the plasma proteome. The plasma proteome has an extraordinary dynamic range: ten orders of magnitude separate the concentration of albumin (40 g/L) from those of the rarest proteins now measured clinically (ng/L levels for some hormones, for example) (Figure 2; Table 1 and Table 2).
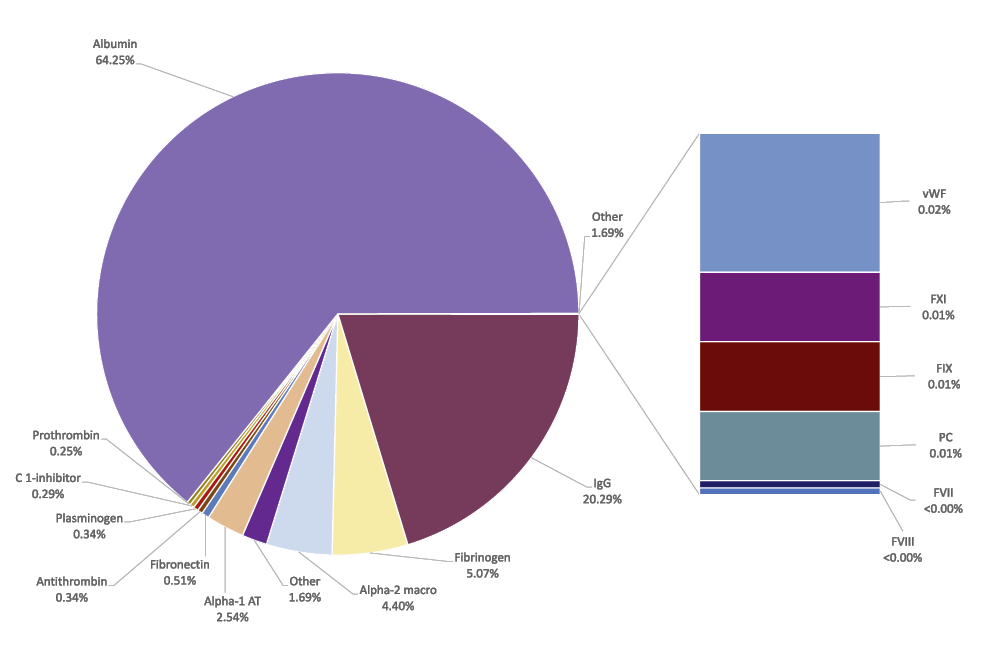
Close to 55 g out of the 60 g protein present in 1 litre of plasma has an established clinical use. (With permission of Prof Th. Burnouf)
The physical properties, dimensions, and size of plasma proteins also differ widely, from 15 micrometres (μm) for albumin (a small molecule with a molecular weight of 66.5 kiloDaltons (kDa) to 90μm for fibrinogen (a huge molecule with a molecular weight of about 340kDa).2
|
Plasma Protein |
Concentration in plasma |
Clinical indication/use |
Estimated disease prevalence/ cases |
|---|---|---|---|
|
Albumin |
40 g/L |
Burns |
|
|
Cardiopulmonary bypass |
|||
|
Cirrhosis complications |
|||
|
Major surgery |
|||
|
Shock |
|||
|
Trauma |
|||
|
Plasma exchange treatments |
|||
|
Acute respiratory distress syndrome |
|||
|
Immunoglobulins |
Up to 12.5 g protein/L |
Chronic inflammatory demyelinating polyneuropathy |
1.5 to 3.6 in 106 people |
|
Acute inflammatory demyelinating polyneuropathy (Guillain-Barré) |
1 to 2 in 100,000 |
||
|
B-cell chronic lymphocytic leukemia |
1 in 200 (US, 2014) |
||
|
Multiple myeloma |
1 in 143 (US) |
||
|
Idiopathic thrombocytopenic purpura |
9.5 in 100,000 |
||
|
Kawasaki disease |
67.3 in 100,000 children under 5 (incidence) |
||
|
Multifocal motor neuropathy |
0.3 in 100,000 (Japan, 2012) |
||
|
Organ and bone marrow transplants |
|||
|
Primary immunodeficiency |
250,000 (US) |
||
|
Cytomegalovirus |
1 in 1,000 births (US, 2011) |
||
|
Hepatitis A |
|||
|
Hepatitis B |
850,000 to 2.2 million people (US) |
||
|
Inhalational anthrax |
|||
|
Rabies |
|||
|
Rh disease |
6 in 1,000 (US) |
||
|
Tetanus |
|||
|
Vaccinia vaccine complications |
3.0 to 38.5 in 106 people (eczema vaccinatum) |
||
|
Varicella |
|
Plasma Protein |
Concentration in plasma |
Clinical indication/use |
Estimated disease prevalence/cases |
|---|---|---|---|
|
Coagulation Factors |
|||
|
Fibrinogen |
3 g/L |
Tissue sealant component |
|
|
Thrombine-Factor II |
150 µg/mL |
Tissue sealant component - Factor II deficiency |
0.33 in 106 to 1 in 107 |
|
Fibronectin |
300 µg/mL |
Wound healing |
|
|
Factor VII |
0.5 µg/mL |
Inhibitors of FVIII and FIX |
0.31 in 106 to 1 in 107 |
|
Factor VIII |
0.5 µg/mL |
Factor VIII deficiency - Haemophilia A |
1 in 10,000 |
|
Factor IX |
10 µg/mL |
Factor IX deficiency - Haemophilia B |
1 in 25,000 |
|
Factor XI |
0.3 µg/mL |
Factor XI deficiency - Haemophilia C |
|
|
Factor XIII |
30 µg/mL |
Factor XIII deficiency |
|
|
von Willebrand Factor |
10 µg/mL |
von Willebrand disease |
1.25 in 106 (US) |
|
Factor V |
0.3 µg/mL |
Factor V deficiency |
1 in 106 |
|
Protease inhibitors |
|||
|
Alpha1-proteinase inhibitor |
1.5 mg/mL |
Hereditary emphysema |
100,000 cases (US) |
|
Antithrombin III |
100 µg/mL |
Antithrombin deficiency |
Between 1 in 20,000 to 1 in 2,000 |
|
C1-inhibitor |
170 µg/mL |
Hereditary angioederma |
1 in 50,000 to 150,000 |
One litre of plasma contains approximately 60 grams of protein, including 55 grams of proteins of established clinical importance. Many of these proteins in concentrated form have been granted designation as medicines for orphan diseases, as they are used for treatment or prevention of rare clinical conditions affecting no more than 5 out of 10,000 people in the European Union.9
In the United States, rare diseases are classified as any disease that affects 1 in 200,000 Americans. Orphan diseases, including rare diseases, are neglected conditions whose treatments are often not considered profitable due to the high costs of the development of medicines and the small size of the patient population.10
Tables 1 and 2 (below) show plasma proteins of current therapeutic interest, their wide range of concentrations in plasma, and the prevalence of diseases where a particular protein is missing or malfunctions.
Currently, about 30 distinct plasma protein products with clinical indications can be isolated from human plasma.11 A number of them, such as immunoglobulin and clotting factors, are included in the WHO Model List of Essential Medicines as essential medicinal products. The WHO urges governments to ensure their citizens can access these medicines, when and where they need them, as a vital step in a country’s progress towards universal health coverage.12,13 Their clinical indications and prospects are reviewed in Strengers14 (see also section 9D.4) and are summarised in Tables 1 and 2.
Polyvalent (i.e. active against more than one antigen) immunoglobulin is needed to treat various immunological disorders such as primary and secondary immune deficiencies (Table 1). In addition, high-dose polyvalent intravenous immunoglobulin has an immune modulating effect (stimulates, suppresses or adapts the immune system) in neurological, haematological, and dermatological immune diseases. Hyper-immune (i.e. with a higher concentration of a specific antibody) immunoglobulin is used to treat several infections, such as measles, tetanus, cytomegalovirus, hepatitis B and rabies, and anti-D immunoglobulin is administered to prevent haemolytic disease of the newborn.
Plasma coagulation concentrates are the treatment of choice for patients with congenital or acquired deficiencies. Anti-protease inhibitors and anticoagulants are used to treat patients suffering from deficiencies (see Table 2). Fibrin sealant and fibrin glue are used as topical haemostatic agents or alongside surgery. Fibrin sealant is a two-component protein preparation combining fibrinogen (with which other proteins might co-purify, such as von Willebrand Factor and fibronectin) and thrombin. Albumin is mainly used as a physiological plasma expander administered in severe conditions. Academic research has further demonstrated that besides maintaining colloid osmotic pressure, albumin plays an important physiological role in the binding and transport of lipids, drugs, hormones, and metabolites, and is a scavenger of naturally generated toxic molecules known as free radicals.15
There are a number of key distinctions between PDMPs and classic pharmaceutical products. Purified plasma proteins used as medicines are referred to by regulatory authorities as ”biologicals” or ”biological therapeutics”. Biologicals are distinguished from other medicines in that they are generally proteins purified from transgenic (genetically modified) animals, from living culture systems or blood, whereas other medicines (considered to be “small molecules”) are either made synthetically or purified from plants.16 Because of these differences, biologicals are subject to separate regulations, tests and controls as well as standard pharmaceutical regulations. The pathogens that may be present in donor blood require that safety is of high interest and of a particular focus.
The volume of plasma used ranges from millilitres for diagnostic applications to thousands of litres for industrial-scale plasma fractionation.
At European level, numerous initiatives related to the blood and plasma sectors had been undertaken prior to the first European Community Directive in 1989 (Directive 89/381/ECC). This Directive was a result of the opening of the single European Market and pursued in cooperation with the Commission of the European Community. With acceptance of EC Directive 89/381, member states reached political agreement regarding the free exchange of goods and services including plasma products after 1992. Furthermore, this Directive implied that member states should aim for self-sufficiency in plasma products in the European Community as a whole, based on the use of voluntary non-remunerated blood donors. Finally, the EC-Directive demanded that all organisations involved in the preparation of plasma products apply standards of Good Manufacturing Practice (GMP) and quality assurance and required all plasma fractionation facilities to have manufacturing and product licences.
Prior to the establishment of legislation by the European Commission, the Council of Europe (Strasbourg, France), in its aim at ensuring safety and availability of blood and its derivatives in a way which respects ethical principles with voluntary and non-remunerated blood donation, had issued recommendations on different aspects of blood transfusion medicine. Examples are the CE Recommendation No. R (81) which recommends that member states establish regulations concerning the importation of blood and blood products, CE Recommendation No. R (88) 4 on the responsibilities of health authorities in the field of blood transfusion, CE Recommendation No. R (90) on plasma products and European self-sufficiency, and CE Recommendation No. R (80) 5 on the use of plasma products for the treatment of haemophilia. In 1977, these Recommendations were drafted by the Committee of Experts on Blood Transfusion and Immunohaematology (SP-HM). The UK was represented in the Committee of Experts by the National Director of the National Blood Transfusion Service or a Director of a Regional Transfusion Centre.
Recommendations are submitted for approval and adoption by the Committee of Ministers of the member states of the Council of Europe. If adopted by the Committee of Ministers, the recommendations are for governments of members to adopt legislation in conformity with the principles appended to the recommendations and take any other measures to ensure their implementation. All previously mentioned recommendations were adopted by the Committee of Ministers including the Minister for the UK.
The UK has not terminated its membership of the Council of Europe. The European Committee on Blood Transfusion (Steering Committee) (CD-P-TS), a commission of the European Directorate for the Quality of Medicines & HealthCare (EDQM) of the Council of Europe, is composed of 46 experts from 38 Council of Europe member states and 12 observers, including the EU Commission and WHO, and meets at least once a year to discuss its work program during the plenary session. CD-P-TS concentrates on studying the ethical, legal and organisational aspects of blood transfusion with a view to ensuring quality, increasing availability, avoiding wastage, ensuring optimal use of blood supplies and analysing the possible ethical and organisational impact of new scientific developments. Currently, the CD-P-TS is chaired by the Director of the Joint United Kingdom Blood Transfusion and Tissue Transplantation Services Professional Advisory Committee (JPAC) at National Blood Service in the UK.
The Council of Europe and the European Commission (EC) are two key organisations working together in the field of substances of human origin, such as blood, tissues, cells and organs, used in a variety of medical therapies with the common, shared goal of protecting public health. The EDQM/Council of Europe sets ethical, safety and quality standards for blood, tissues, cells and organs. Through its programs and legal instruments, it works to ensure the quality and safety of blood transfusions in the 47 member states of the Council of Europe and beyond. The European Commission/DG SANTE undertakes a range of activities, including drafting legislation and developing guidance, assisting national authorities with their implementation, accompanied by vigilance activities and project support for its 27 member states. These two organisations co-ordinate their efforts and resources to avoid any overlap or gap in existing regulations in the field of blood and blood components.17,18
The legal requirements specifically for blood and plasma are formulated on a European level in directives of the European Commission. The competent national authorities adopt these requirements. These directives include standards of quality and safety for the collection, testing, processing, storage and distribution of blood and blood components, as well as requirements in relation to traceability and the notification of serious adverse reactions and events. The directives also include standards and specifications relating to a quality control system for organisations providing blood services.19,20,21,22,23,24 These standards are still applied in the UK.
The history of concentrated plasma derived medicinal products such as clotting factor concentrates, immunoglobulin preparations, albumin and other products begins in the early 1940s, when Dr. Edwin J. Cohn pioneered fractionation of plasma with various proportions of alcohol. Using plasma obtained from blood and plasma donors as the source material, fractions from the manufacturing process were separated and these fractions contained concentrated plasma proteins. Twenty years later, in 1964, Dr. Judith Pool and coworkers were able to make another brilliant discovery by observing that when pooled plasma, frozen in large containers, was thawed cautiously it left a small amount of unthawed fibrous-looking paste at the bottom of the containers (cryoprecipitate). By testing this paste, they recognised that Factor VIII was concentrated in the cryoprecipitate.
Plasma fractionation starts at the time the donation is collected from the donor. Ensuring the safety and quality of plasma products relies on a complex system starting with careful donor selection and testing.25
Blood and plasma donations are obtained from healthy volunteer donors and constitute a very generous gift particularly in the current context of the Covid-19 crisis (see Figure 2).26
In its aim of ensuring safety and availability of blood and its derivatives in a way which respects ethical principles, the Council of Europe (Strasbourg, France) has issued recommendations on different aspects of blood transfusion medicine. The basis for this ethical principle is that the donation is voluntary and non-remunerated. This principle is also promoted by the World Health Organisation, the International Red Cross and Red Crescent Societies, and the International Society of Blood Transfusion. Regarding the safety of blood and plasma on the transmission of blood-borne pathogens, this principle became more paramount given the evidence that paid blood and plasma and plasma obtained from involuntary donors such as inmates had a higher risk of being infected with transfusion-transmitted infections such as hepatitis B and HIV.
Donors are first informed and screened by interview, using an extended questionnaire, and by a physical examination to detect obvious risk factors and deficiencies. This is the first step that determines donor inclusion or exclusion, ensuring that high-risk donors are excluded from donating.
Each parameter should be optimised: donor information, donor selection, donor testing, plasma or whole blood collection time, type and concentration of anticoagulant in the collection bag, method of storage, rate of cooling, time of separation of plasma from cellular elements, time and duration of centrifugation, plasma storage, and the rate of freezing and thawing.27,28 Different tests are carried out on each single donation to detect the presence of viruses, bacteria or other pathogens that can be transmitted by blood, depending on the donation time period and local epidemiology, and on national and international regulations.29
Today, all donations must be tested for human immunodeficiency virus (HIV1 and HIV2 antigens, antibodies, or both), hepatitis B virus (HBsAg), hepatitis C virus (antigens and antibodies), and syphilis.
Plasma donations can be classified according to the method of collection. Plasma can be “recovered” as a by-product of whole blood processing into cellular components and plasma, or obtained as “source plasma” by apheresis. This latter approach, known as plasmapheresis, is a process by which during the donation procedure, a machine removes the red cells and platelets from whole blood, and returns the cells to the donor while retaining the plasma. Both recovered and source plasma are appropriate for use as starting material for the manufacture of plasma derivatives.30 Concurrent plasma, a by-product collected during the apheresis collection procedure used to obtain platelets (plateletpheresis), is also suitable for further fractionation.31 The entire plasma donation process by using the apheresis procedure takes approximately 90-120 minutes.
Most source plasma comes from paid plasma donors in the US, Austria, the Czech Republic, Germany, Hungary, Ukraine, and China. Plasma collected from paid donations in the US provides 65-70% of the global volume of source plasma, and Europe is currently reliant on the US for 37% of the plasma it uses.
The maximum volume of plasma that can be donated, and the donation frequency are regulated by national authorities. For recovered plasma, the authorised volume ranges from 450 (±10%) to 500 (±10%) ml per donation, anticoagulant excluded. The figure of 450 ml relates to the total blood donation, of which a little more than 50% is plasma. A unit of recovered plasma can be used as fresh frozen plasma or plasma for transfusion in hospitals, as a source of cryoprecipitate, for convalescent plasma therapy (as it was for early COVID-19 patients because of its high COVID-19 antibody titre), or as a source to manufacture PDMPs.
For apheresis plasma, the authorised volume ranges from 400 to 800 ml/donation, anticoagulant excluded. Donation frequency ranges from 3 to 5 times a year for whole blood and from 15 to 104 times a year for plasma. Differences in plasma protein levels can depend on the plasma collection technique and frequency of donation (for example, more Factor VIII and less immunoglobulin can be found in plasma with more frequent collections/time period).32,33,34,35 Compared to the collection of recovered plasma, plasmapheresis allows larger annual plasma volumes per donor to be made available for fractionation, thanks to a combination of higher donation frequency and larger volume per donation.
In the US a study of donor demographic data for 2012, involving 15 million donors and 25.2 million plasmapheresis donations from seven participating companies, indicated that the average number of donations per donor was 17.3, with 49% of donors having donated fewer than 7 times and 9.8% more than 50 times.36 70% of the donors had been donating for two years or less, indicating the need to recruit new donors constantly.
There are significant regional differences in collected volumes of recovered and source plasma. In 2017, the US supplied 65-70% of the world supply of plasma for fractionation, whereas Europe was the largest supplier of recovered plasma, with only 10% of the source plasma. Latin America and Africa currently account for a small proportion of the global plasma supply but have a rapidly growing demand for plasma products.37
Over the time period of 1970-2000, the impact of paid or remunerated plasma donations has changed. One of the main arguments for self-sufficiency was that blood from paid donors was more often infected with hepatitis virus than blood from voluntary non-remunerated donors. Comparative studies published between 1968 and 2001 were assessed for a possible trend of change in the relative risk for infectious disease markers between paid and unpaid blood or plasma donors.38 Studies reporting that paid donors had lower risk were found, but most studies continued to report that paid donors have higher rates of infectious disease markers than unpaid donors. Paid donors were still more likely than unpaid donors to donate blood in the period during which infectious donations escape detection by blood-screening tests (the “window-period”). The risk of being contaminated was much smaller for cryoprecipitate made from a small number of donation units than for plasma concentrates from pooled plasma.
The introduction of additional safety measures for handling plasma donations and the preparation, purification and viral-inactivation steps employed for the production of plasma derivatives have significantly improved this unfortunate situation.39
There is no evidence from clinical studies or pharmacovigilance that donor remuneration increases the risk of viral transmission via PDMPs when proper screening is carried out at donation, each unit is tested according to the latest requirements and when validated virus inactivation/removal steps are applied during manufacture.40
There is a delicate balance between plasma supply and demand. Demand for PDMPs is steadily increasing, driven by global increased access to medical care, new products and applications, and diagnostic advances.41 Experience has shown that national sources of plasma can become unusable from one day to the next, as happened during the variant Creutzfeldt-Jakob disease crisis in the UK in the 1990s (see section 9B.8.v). More recently in 2019-2021, intravenous immunoglobulin (IVIg) was short in supply. With the COVID-19 pandemic, the global plasma supply has come under additional pressure.
There is an emphasis on maximising the use of the recovered plasma collected for fractionation. In 2021, the WHO guidance on “Increased supplies of plasma-derived medicinal products in low- and middle-income countries through fractionation of domestic plasma” encouraged member states to increase their volume of quality plasma for fractionation to allow for better treatment of patients who need PDMPs.42 In low- and middle-income countries, millions of litres of recovered plasma (plasma separated from whole blood into components) are discarded as waste because the quality assurance, technology, infrastructure and regulatory oversight required to ensure the quality of the plasma for fractionation are lacking.
In 2021, people in the UK are permitted to donate plasma for medicines. The plasma is intended for use to manufacture immunoglobulins.43 (See also section 9B.8.v).
Nevertheless, for the World Health Organisation, voluntary non-remunerated blood donations from low risk populations remains the foundation of safe, adequate and sustainable blood supply for transfusion that also can support patient needs for therapies with PDMPs.44,45
Plasma may have different qualities regarding the protein content and the concentrations of clotting factors. The normal albumin level ranges between 35-55 g/L. The normal immunoglobulin level ranges between 7-16 g/L and depends on race, epidemiology of endogenous diseases and location. Since the half-life of endogenous immunoglobulin is around 3 weeks, an interval of less than 3 weeks between two plasma donations may negatively influence the remaining immunoglobulin level in the plasma of the donor.
The normal blood level for the clotting factor Factor VIII is 100% with a range of 60-150% according to different factors such as genetic and environmental factors, blood group, disease and stress response to injury. Since Factor VIII is an acute phase protein, the concentration of Factor VIII in the blood is influenced by the way blood is drawn from the donor, the storage temperature and the handling of the donation. A low blood flow and too high a temperature will lower the plasma level of Factor VIII in the blood bag.
Plasma is usually shipped frozen in individual units from local blood or plasma collection centres to a central processing plant.
The first step in the production process in a manufacturing plant is to identify each donation to guarantee traceability and to examine the integrity of the frozen plasma bags, before transferring the contents of the bag to the first thawing tank (holding 100 to 10,000 donations depending on the manufacturing procedure) and crushing it. The whole volume of plasma is thawed and then the precipitate collected in one step or, preferably continuous thawing is implemented allowing the cryoprecipitate to be continuously separated from the liquid phase. The first homogeneous mixture, the “plasma pool”, is obtained after collecting the cryoprecipitate or any other intermediate.
To ensure the safety of the plasma-derived products, plasma pools are tested for the presence of hepatitis C by Nucleic Acid Amplification Testing (e.g. PCR). An overview of the testing process mandatorily required by UK authorities for five viruses (hepatitis C virus, hepatitis A virus, and parvovirus B19 by nucleic acid amplification, human immunodeficiency virus by serology, and hepatitis B by antigen detection) can be found on the National Institute for Biological Standards and Control website.46
Today, fractionators must define precisely the quality and the safety of plasma donations in terms of specifications for collection, testing, storage, and distribution. The Plasma Master File, which requires approval by the European Medicines Agency (EMA) lists these manufacturing specifications for Europe.47 It is included in the medicines’ dossier and approved by the Regulatory Agencies such as the EMA and the UK Medicines and Healthcare products Regulatory Agency (MHRA).48 This file must be compliant with the relevant European Pharmacopeia 6.2. Monograph 07/2008:0853,49 and to guideline EMEA/CPMP/BWP/3794/03.50
The following section outlines the different fractionation processes developed by Dr E.J. Cohn including his initial research just before and during World War II. It goes on to set out the technical aspects of the Cohn fractionation process, and ends with a discussion of the safety aspects related in particular to hepatitis.
Separation of the many protein and lipid components in a biological fluid can be carried out by controlling their relative solubilities in a multivariable system.51 The purification of plasma proteins relies on the fact that a protein is differently charged in alkaline and acidic solutions and is neutral (carries no net charge) at the pH called its isoelectric point. The isoelectric point of each protein is specific to that protein. Proteins are less soluble at or near the isoelectric point and might then precipitate (become solid)52 or flocculate (form small clumps).53 This is specific to each type of protein. For instance, globulins such as immunoglobulin, an important class of proteins, become soluble when a small amount of salt is added but may precipitate at a higher salt concentration (“salting-out”). Thus, precipitation may not be able to solve all purification challenges but can be used to remove contaminants or to precipitate target proteins.54
Protein concentration is also a crucial parameter. Plasma proteins are generally far more soluble in aqueous solution than in organic liquids such as alcohol. The addition of alcohol exerts an extra powerful precipitating effect by reducing protein polarity (electrostatic interactions between groups on the protein surface) and should be performed at low temperature to avoid protein alteration or denaturation.55 The temperature to be used depends on the alcohol concentration (in this situation the ethanol concentration) and should be low enough to avoid protein alteration. This procedure reduces the danger of bacterial growth. It is also possible to vary the temperature to effect reproducible separations.
When ethanol is removed, the presence of neutral salts will increase the solubility of globulins in water.
Precipitation is a robust method and quite inexpensive. Today, predictive modelling of the impact of precipitation is possible on the basis of protein primary structure.56
Figure 3 overleaf shows the milestones of fractionation and viral safety (tests, virus inactivation/removal methods during manufacturing, regulations) from the 1940s to the 2000s.
Figure 3 overleaf describes the parallel development of the production of new therapeutical PDMPs (See also Figures 4 to 6), starting from blood plasma, used to treat different specific diseases as soon as the medical knowledge evolved.
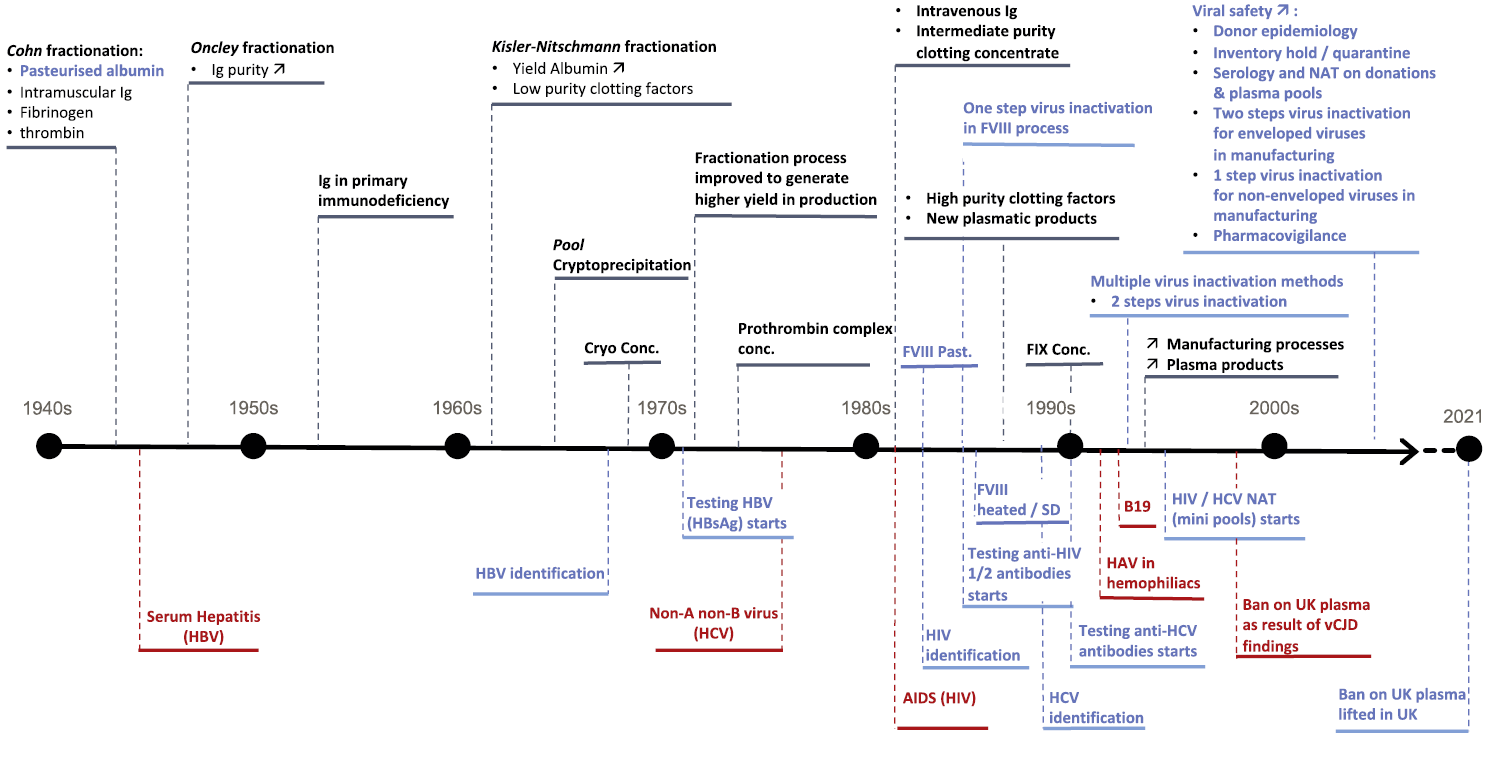
Fractionation evolution is in black. Recognised disease events are in red. Contribution to safety is in blue.
Since the beginning of plasma fractionation, there was a concern that existing and new emerging blood-borne pathogens may enter the blood supply and threaten the safety of plasma products. As soon as an infectious disease was recognised in PDMP-treated patients, and the pathogen agent could be identified, adequate safety measures were proposed.
The emergence and the identification of hepatitis B, AIDS, hepatitis C, vCJD and new pathogenic viruses (see also table 4) that were transmitted by therapeutic PDMPs, had a powerful influence on:
The procedure for purifying plasma proteins of high therapeutic interest that continues to play a pivotal role in plasma fractionation was developed by Dr Edwin Cohn following his research at the Department of Physical Chemistry, Harvard Medical School. The history of industrial plasma fractionation began in about 1938, when Cohn and colleagues undertook a systematic study of the fractionation of blood plasma proteins.57 Cohn emphasised from the beginning that blood plasma contains many protein constituents, each with its specific function, and that not using all plasma constituents for the treatment of patients with specific plasma-component needs is a waste of precious material.58 His goal was to develop an inclusive method by which all plasma constituents could be preserved and made available in purified form. At that time, albumin was by far the most effective fraction for the treatment of patients in shock who needed a protein solution to restore depleted plasma volume. Albumin is an essential protein for maintaining the osmotic pressure between the intravascular system and other body tissues. For patients requiring immunisation against certain diseases, immunoglobulin could be the required fraction as it contains most antibodies.
In the spring of 1940, when medical resources first began to be mobilised with the realisation that the US would likely enter World War II, the use of blood serum for illnesses and other conditions was limited to a small number of pioneer researchers in a few medical centres. The technique of rapidly freezing protein solutions to very low temperatures and then drying them in vacuum from the frozen state had recently been developed allowing the use of large quantities of protein.59 In 1941, freeze-dried plasma was commercialised in bottles of material equivalent to 250 ml or 500 ml, ready to be reconstituted with companion bottles of distilled water. The use of human whole blood and later freeze-dried plasma was known to be associated with a risk of causing transfusion reactions, immunisations, and transfer of diseases, particularly hepatitis.60,61 In the autumn of 1942, epidemic hepatitis was increasingly associated with transfusions of whole blood and plasma,62 but with the administration of plasma compounds these risks were either diminished or completely avoided and, because of their high concentration, therapeutic efficacy increased.63 Moreover, these plasma compounds were easier to ship because of the smaller volume in the final container. It was hoped at that time that bovine albumin could be prepared and used as a plasma expander for the treatment of patients in shock but, as Cohn suspected, it proved not to be safe for use in humans. This led to Cohn studying the purification of albumin from human plasma.
The development of large-scale methods for preparing therapeutic human proteins was enormously accelerated by the outbreak of World War II. The US Navy requested 300,000 units of human whole blood or plasma, a very large amount at the time.64 Considering that albumin could be used instead of plasma, Cohn and colleagues developed a continuous system for separating plasma proteins into five major fractions called (in order of the successive production steps) Fraction I, Fraction II, Fraction III, Fraction IV, and Fraction V. This system relied on controlling the relative solubility of these fractions in a multivariable system.65 When war broke out in December 1941, Cohn’s laboratory shipped human serum albumin (processed Fraction V) using plasma from the Red Cross to treat casualties of the Pearl Harbour attack. A small number of severely burned patients were given albumin and all showed prompt clinical improvement.66 Albumin continued to be the highest-priority product, notably in 1944 during the Allied invasion of Normandy, but other biologicals were prepared for civilian and military medicine. According to reports, purified immunoglobulin was transfused to make children immune to measles and was thought to be safe.67,68,69 Starting from a pool of convalescent plasma, concentrations of specific antibodies reached up to 25 times those produced from pools of normal plasma.
The fractionation of plasma proteins was carried out for the Armed Forces with blood from two million donors.70 Up to 1945, about 14 tons of albumin (25g/bottle) was provided by seven companies (Lederle, Lilly, Squibb, Cutter, Sharp & Dohme, Upjohn, and Armour). In 1945, Cohn submitted a report describing the isotonic liquid preparation of albumin (25%) stabilised with acetyl tryptophan or mandelic acid, filtered through non-absolute asbestos pads, and pasteurised.71
Cohn fractionation is a technology that guarantees reproducible preparation of components for the preparation of plasma derivatives for clinical use.
The safety of plasma preparations had to be guaranteed by avoiding possible modification of the native structure of protein(s) and contamination by undesirable proteins or pathogens causing side effects such as aggregate formation, thrombogenicity, haemolysis, pyrogenicity, immunosuppressive effects, immunogenicity, or infection.72
The purpose of the ethanol fractionation process is to fractionate plasma, i.e. to divide plasma into a series of fractions, each containing a particular therapeutic protein at a variable concentration and degree of purity.
Protein separation is performed under conditions where the solubility of the protein of interest is maximised and that of the other proteins is minimised or nil. Manipulation of five different variables allows maximum separation of proteins.73 This multi-step procedure is suitable for purifying the major proteins present in the starting material.
The use of ethanol, a cheap precipitant, allows the process to operate at low temperature and minimises bacterial growth. More recently developed methods of desiccation i.e. freeze-drying allow the prolonged storage of fractions, some of which are not stable in solution, in a powdered state. Before use, the powder is reconstituted with sterile distilled water.
The starting material for manufacturing is the plasma pool which is prepared prior to plasma fractionation by mixing many plasma donation units (often 1,000 and sometimes up to 10,000 or more) each of which is collected by plasmapheresis or separated from whole blood.
The size of a plasma pool is determined as a minimum of 1,000 units of plasma, because the manufacturing of immunoglobulin preparations requires a broad spectrum of antibodies from many donors to be present in the final product for the treatment of immune deficient patients. However, as manufactures increased the size of the plasma pool with higher numbers of units, for economic reasons, papers were published in the 1990s warning that the risk of exposure to transfusion transmitted infections increases with the pool size and the prevalence of the transfusion transmitted agent, and accumulates with repeated treatments with material manufactured from different pools.74
The parameters used for precipitation of target proteins are:
From the beginning, Cohn and his co-workers made changes to cold ethanol precipitation, a step which continues to be modified and adapted by different fractionation centres. Nevertheless, all these subsequent improvements align with the main principles.
Of the ten different fractionation methods (not including variations) developed using this strategy, the three most used are Cohn method 6, Cohn method 9 modified according to Oncley et al.75 and Cohn method 6 modified according to Kistler and Nitschmann.76
Cohn assessed the success of separation on the basis of three criteria:
Cohn also stressed the purity and concentration of the component of therapeutic value to obtain high potency.
In this process variant, there are four fractions of interest. Fractions I, II+III, and IV are obtained after successive precipitation steps in the presence of 8%, 25%, and 40% ethanol respectively (Figure 4). The pH varies from 7.2 to 4.6. The temperature is decreased in parallel with the ethanol concentration from -3°C to -7°C to avoid denaturation and aggregation. The procedure also depends on low ionic strength, accurate control of pH and the protein concentration.
At each step, the solid precipitate is collected by semi-continuous centrifugation and the liquid supernatant (the liquid remaining on the solid residue) is further processed by adjusting the pH, ionic strength, protein concentration, and ethanol concentration.77,78,79,80
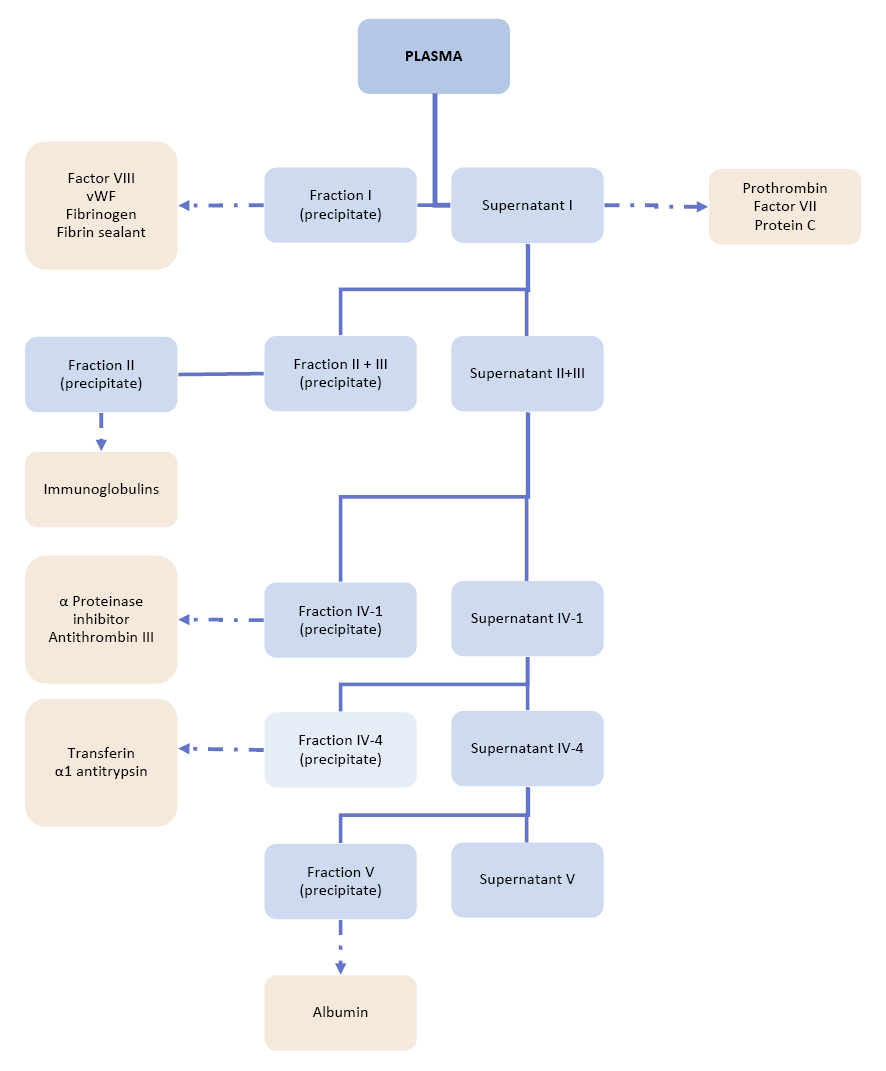
Plasma is fractionated into five successive fractions (from I to V), by modifying the ionic strength, pH, ethanol concentration, temperature and adjusting protein concentration.
Fraction I (5-10% of the initial plasma protein) contains most of the fibrinogen, von Willebrand Factor, and anti-haemophilic Factor (anti-haemophilic Factor or Factor VIII or FVIII are different names for the same clotting factor) (circa 50% of clottable proteins; all huge molecules), and it is obtained after precipitation at 8% ethanol and pH 7.2, ionic strength 0.14, -3°C, protein 5.1%. Fraction I as a clotting factor source was always produced from small plasma pools for haemophilia treatment originally.
When in the early 1940s, Cohn pioneered fractionation of plasma with various proportions of ethanol, his ‘fraction I’ contained mostly fibrinogen and also Factor VIII, but the methods of testing the concentration by using a Factor VIII assay had not yet been developed. The utility of fraction I in haemophilia treatment was demonstrated in 1947 and modest amounts were used in developed countries throughout the 1950s and 1960s. An early Factor VIII production process (Cohn Fraction I) was developed in 1956 in Scotland. From 1956 to the second half of 1980s, a preparation derived from Fraction I was produced under the name of I-O (later named AHF-Kabi when commercially produced) in Sweden and the Nordic countries. The product was withdrawn notably because of insufficient virus inactivation (see section 9B.5).81,82
Discovery of its clotting capacity preceded that of the clotting cascade reaction and characterisation of all the clotting factors. Fraction I also contains small molecules of the complement system (the part of the immune system that enhances the function of antibodies) and cold insoluble globulins. Supernatant I is processed further by increasing the ethanol concentration.
Fraction II + III (25% of the initial plasma protein) obtained after precipitation of Supernatant I at 25% ethanol (pH 6.9, ionic strength 0.09, -5°C), contains immunoglobulin (IgG, IgA, IgM), clotting factors (Factor II, Factor VII, Factor IX, Factor X), and α- and β-globulins). It is further fractionated by serial ethanol precipitations to yield Fraction II, containing immunoglobulin of early proven value, as in the prophylaxis of measles. Purity reaches 85%.
Supernatant II+III is in turn subjected to precipitation in the presence of 18% ethanol, (pH 5.2, ionic strength 0.09, -5°C, protein 3%), yielding Fraction IV-1 (5-10% of the initial plasma protein) containing α- and β-globulins, IgM, antithrombin III, and complement components. By increasing the ethanol concentration in the supernatant to 40%, Fraction IV-4 is obtained, containing 5-10% of the initial protein and rich in α- and β-globulins, ceruloplasmin, haptoglobin, and transferrin (ionic strength 0.11, -5°C°, protein 1%).
Fraction V is finally obtained from supernatant IV-4 containing 40% ethanol, by decreasing the pH to 4.8, near the isoelectric point 4.9 of albumin (ionic strength 0.11, -5°C, protein 0.8%) and then collecting the precipitate. Fraction V contains 85 to 98% albumin, a small molecule remarkably soluble under these extreme conditions.
The final processing steps include depth filtration, formulation, stabilisation, and a final filtration. For albumin, an additional final pasteurisation in its final container was performed in the 1940s. The human albumin molecule has many binding sites for various molecules and drugs.83 By filling these molecular sites with caprylate or mandelate (both of which have antimicrobial activity), one stabilises the albumin conformational structure during pasteurisation.
It must be stressed that until the 1970s, albumin and intramuscular immunoglobulin (in low volumes) were the most important products of fractionation. Interestingly, even in 1975, Schneider et al. did not suspect another purpose for Fraction II other than low-demand intramuscular immunoglobulin.
When ethanol fractionation is applied to plasma, fibrinogen (the precursor of fibrin, the structural element of blood clots) is concentrated in the first fraction “Fraction I”.
Fraction II+III contains prothrombin, which, after purification and conversion to thrombin, can be made available as a dry powder ready for reconstitution with sterile water, to convert fibrinogen to fibrin. Fibrin film displays unique properties and can be effective as a haemostatic agent for immediate emergency use.84
Albumin, immune globulins, fibrin foam, and thrombin were licensed under the US National Institute of Health and were prepared in large amounts under a contract with the US Navy in June 1944.85
The Cohn procedure can undergo many modifications to provide a range of fractions rich in the protein(s) desired. Cohn et al developed different methods, but Method 6 is the one most used in Europe and North America, with the Cohn alcohol precipitation system based on Cohn method 6 adopted in Edinburgh in 1951 on a modest scale.86 To increase albumin recovery, improve productivity or facilitate working schedules, manufacturers introduced ”in house” modifications of these initial processes by slightly modifying parameters such as pH, osmolarity, and contact time with ethanol. They also introduced direct ultrafiltration of Fraction IV-1 when this technology became available. These adaptations were implemented mostly to increase albumin recovery or to limit night shifts or manual production steps over the weekend.87
In the first variant of the process, the Fraction V paste (crude or refined) was lyophilised to remove the ethanol. The resulting powder was then dissolved, the solution clarified by filtration with depth filters in the presence of filter aids, and the pH and osmolarity adjusted.88 Today, extensive ultrafiltration/diafiltration is performed instead of freeze-drying. The protein concentration is adjusted to 4% or to 20-25% if a higher-concentration albumin product is required. The addition of caprylate (a fatty acid with antimicrobial properties) stabilises the albumin molecule and allows, after sterile depth filtration and dispensing into sterile bottles, pasteurisation in the final container (60°C – 10 h) to ensure a high level of safety. Depending on the manufacturer, acetyltryptophan or mandelate is also added along with caprylate for this final pasteurisation.
Albumin purity is of high importance. The product should be free of contamination with other proteins, endotoxins, metal ions, aggregates, and prekallikrein activators. A comparison of different process variants and of their continuous improvement is described by Matejtschuk et al.89
Because of the presence of ethanol, the entire large-scale fractionation process must take place in a cold room or in self-cooling machines, and in a pharmaceutical environment.
In the period around World War II, it became known that patients could get jaundice. In 1942, it was recognised that 23,000 American soldiers (with 62 deaths) got jaundice after being vaccinated with a yellow fever vaccine which was stabilised with human plasma.90 Now we know that this plasma was contaminated with hepatitis B virus.91 As thought in the 1940s, the paediatrician Krugman determined in the fifties and sixties in the US – by contamination studies in children – that there were at least two types of contagious jaundice: one type with a short and one with a long incubation time. Later it turned out that these were hepatitis A and B. At the beginning of the 1960s the American Nobel prize winner Blumberg discovered the so called ‘Australian antigen’, later connected (by Prince) with HBV. Subsequently in the 1970s, tests were developed to determine HBV-antigens (HBsAg, HBeAg) and antibodies against these antigens (anti-HBs, anti-HBc, and anti-HBe). Shortly thereafter, the hepatitis A virus (HAV) was discovered and diagnostic tests have been developed in order to determine recent (anti-HAV-IgM) and passed-through infections (anti-HAV-IgG). Unfortunately, still an important proportion (2-10%) of patients who suffered after transfusion of blood products from a type of hepatitis for which there was no connection with HBV, HAV, and other hepatotropic viruses such as cytomegalovirus or Epstein-Barr virus. The earlier mentioned Prince called this form of hepatitis ‘hepatitis C’. The name ‘non-A-non-B’ (NANB) was preferred however.
Hepatitis transmission was a source of great concern in relation to albumin prepared from large plasma pools. Laboratory investigations were run on albumin samples spiked with plasma proven on repeated occasions to contain hepatitis virus.92 Owing to the lack of known susceptible laboratory animals, a limited number of human volunteers had to be inoculated in order to demonstrate the presence of active hepatitis. When viruses were added to a small pool of plasma, they survived small-scale fractionation and were found in the non-pasteurised fractions. In the pasteurised fractions they were not found to be present. The results showed the efficacy of final pasteurisation.
Heating albumin, a remarkably thermostable protein, at 60°C for 10 h was successful at eliminating viruses in the presence of acetyltryptophan and caprylate, two added molecules that bind albumin and protect it from denaturation. Unfortunately, with these particular process conditions, it was not possible to apply the methodology to other more labile plasma proteins. The presence of acetyltryptophan, caprylate or a mix of both products do not stabilise the Factor VIII molecule during the heating process.
This pasteurisation procedure, still in use today, was a huge step towards safety with regard to aggregates, bacteria, and particularly viruses.
The value of immunoglobulin-containing Cohn Fraction II in the prevention and attenuation of infectious hepatitis was recognised and the fraction was thought not to transmit serum hepatitis (hepatitis B) to patients.93,94,95,96,97 As a result of this success, a large-scale trial was conducted on World War II forces in the Mediterranean where the disease was epidemic.98 The test was carried out on four squadrons of about 500 men each. Two squadrons were injected intramuscularly and no jaundice cases were detected after 9 weeks in contrast to the two control groups (4 cases and 21 cases).
As a number of plasma products administered to humans were contaminated with viruses, in 1995 the European Agency for the Evaluation of Medicinal Products pointed out the need for and the contribution of virus validation studies to the viral safety of biological products (this topic will be discussed below).99 As a result, every PDMP manufacturer has validated its PDMP production processes using the same rules.
Despite the differences in the process flow-charts used by seven commercial fractionation companies (Biotest, Octapharma, Kedrion, CSL Behring Bern, CSL Behring Marbourg, Baxter BioScience and Talecris), the results of a total of 615 studies were collected, analysed and published in 2011 to study potential virus removal through Cohn fractionation.100
In brief, each fractionation company collected fractions at different steps of the ethanol fractionation during production. Scaled-down processes were run in the presence of the relevant HIV and model viruses for enveloped and non-enveloped viruses, in particular for HBV and HIV, using identical conditions. These viral validation studies were performed in certified virology laboratories. The competent authorities evaluated all the experiments individually. Despite differences in the different production processes, the results show the importance of Cohn fractionation steps for virus removal from cold ethanol fractionation, particularly removal of Fraction III from Fraction II+III or Fraction I+II+III (Fraction II contains nearly all immunoglobulin G), or Fraction IV (the fractionation step before obtaining albumin). Higher ethanol concentration (40%) can contribute to safety through the inactivation of HIV and the viruses used as a model for HBV but not those for HCV. Differences in efficacy could be observed between manufacturers, maybe because of their specific fractionation processes.
In summary, the efficacy of virus removal is moderate for model viruses of HCV, demonstrating that Cohn fractionation is not sufficient to render a final plasma product virus safe.
The following section sets out how fractionation processes developed from the 1960s to the 1990s. It includes a short section on clotting products and detailed explanations of the therapeutic uses of immunoglobulin.
The need for plasma fractions for the manufacture of PDMPs has become the main driving force in plasma collection for all national blood transfusion services and commercial plasma collection centres. New products and new equipment have been developed, and new facilities, either for-profit or not-for-profit, have been built to provide more products and new therapeutic proteins, as they are often the only available option to treat life-threatening conditions.
Cohn’s strategy offers numerous industrial advantages, including common validation steps for these different medicines, reduced development costs (as each intermediate ethanol fraction can be used as the first step for further purification using more flexible technology) and reduced production costs including those related to human resources and in-process quality control. Lastly, these processes have been approved and the products registered, steps which can prove obstacles when implementing totally new technology.
It was estimated that in 1982 approximately 10 million litres of plasma were fractionated worldwide.101 In the decade between 1982 and 1992 the plasma BPL received for manufacturing from the Regional Transfusion Centres in England and Wales increased from 123 tonnes (1982/83) to 490 tonnes (1990/91).102
Clotting factors, in particular anti-haemophilic factors, were increasingly produced from small pools (5-12 bags) to treat haemophilia patients.103 Fibrinogen and anti-haemophilic factor (Cohn Fraction I) prepared from large pools (> 12 bags) was considered less desirable because of detection of the Australian antigen (associated with the hepatitis B virus) and the extremely high risk of hepatitis transmission (see below).104,105 Cohn Fraction I obtained by precipitation with 8% ethanol was produced from small plasma pools made from plasma separated from whole blood within 6 hours of collection. Fraction I produced with the Cohn method was unstable. Since the 1950s, various processes of Fraction I allowed its use after being treated using glycine, ethanol and citrate or using “Synthamin”.
According to Pool et al and Pool and Shannon, the cryoprecipitation production was as follows: thawing of the frozen plasma (2-4° C), centrifugation of the cryoprecipitate, followed by the ethanol fractionation.106,107 The new clotting Factor VIII concentrate was found in the cryoprecipitate. Factor IX was obtained in the cryosupernatant (Figure 4a).
The Factor VIII concentrate was, in terms of recovery of coagulant activity, inferior to the Cohn fraction I but could be prepared with little capital investment.108,109,110
In section 9B.9, an outline will be given on the development and production of Factor VIII products over time and the impact on the availability, safety and efficacy of the substitution treatment for people with haemophilia.
When the demand for albumin declined because of the introduction of gelatins, dextrans and hydroxyethyl starches as plasma volume expanders, Factor VIII became the leading plasma protein which determined the demand for plasma. The demand for FVIII products increased due to better and more efficient FVIII products, new therapeutic interventions in the area of orthopaedic reconstructive surgery, the advocacy role of parent/patient organisations, and the development of comprehensive home care programmes, which were initiated with cryoprecipitate and further developed when the fractionation technology advanced to a point that lyophilised Factor VIII became available. Guidelines for haemophilia treatment were developed and nurse-training programmes were initiated.111
Immunoglobulin refers to a group of closely related glycoproteins present in the plasma at a mean concentration ranging from 7 to 12 g/L depending on individual variations in humans. Immunoglobulin proteins are large glycoproteins with a total mass of approximately 150-190 kDa. All immunoglobulin molecules have the same basic core structure.112 The five primary classes of immunoglobulins are immunoglobulin G (IgG), immunoglobulin M (IgM), immunoglobulin A (IgA), immunoglobulin D (IgD), and immunoglobulin E (IgE). Immunoglobulin G (IgG), a major effector molecule of the humoral immune response (protecting against extra-cellular pathogens) in humans, accounts for about 75% of the total immunoglobulin in the plasma of healthy individuals. None of these classes of immunoglobulin represents a homogeneous population of molecules which differ in physical, chemical, immunological and other biological properties.
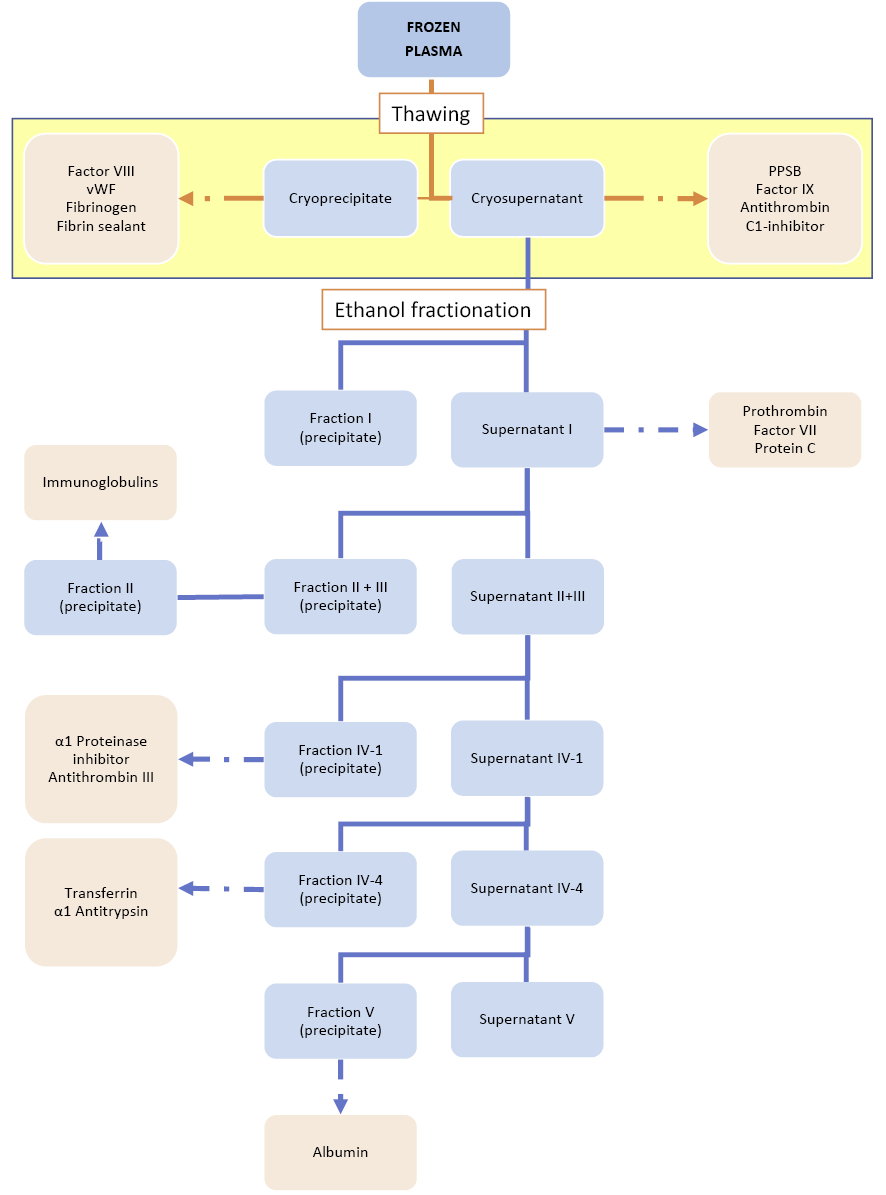
After thawing the plasma, the cryoprecipitate is collected. Using the cryosupernatant as the starting material, the ethanol fractionation is processed as in Figure 4.
Preparations obtained from random pools of human plasma may be referred to as “human normal immunoglobulin”.113 This designation corresponds to names such as “gamma globulins”, “normal gamma globulins” or “immune serum globulin”. The literature references these different names to refer to the same protein.
Human immunoglobulin is used in both prophylaxis and on-demand treatment to provide passive immunity through the specific antibodies it contains. The first immunoglobulin products were mainly given to prevent and treat infections: poliomyelitis, measles, mumps, pertussis and Hepatitis A. These specific immunoglobulin products were less used in the developed world once the respective diseases could be prevented by vaccination.114
The production procedures described concern all immunoglobulin products. These include products that are prepared from pools of plasma from non-selected donors, and products that are prepared from pools of plasma from selected donors who have high antibody titers against specific antigens. These high titers are developed by natural immunisation after disease or by immunisation after vaccination.
After local virus infection outbreaks in the donor population or following epidemics such as measles, hepatitis A or COVID-19, or after specific immunisation procedures (e.g. against diphtheria or tetanus), the collected plasma becomes relatively rich in antibodies against these pathogens. The donor plasma units rich in specific antibodies are the starting material for the specific antibody preparations, said to be hyperimmune because they contain a high titre of specific antibodies against a virus or a specific antigen (such as anti-D antibodies) as defined by laboratory determination.
The concentration of these specific antibodies should not be below those indicated in the European Pharmacopoeia.
The demands for hyper immune immunoglobulin products for various applications (preventing rhesus immunisation, antibodies with specific activity against the tetanus toxin, antibodies against the vaccinia virus to prevent infection and others indications) have created a new need to expand immunoglobulin production.115
A polyvalent immunoglobulin, i.e. active against several antigens, contains all of the antibody specificities found in a plasma pool which is produced from more than 1,000 donations of human plasma obtained from healthy blood and plasma donors. The product is originally in use for antibody replacement therapy, but the use has been extended to other clinical conditions due to their anti-inflammatory and immunomodulatory effects. The latter was not anticipated when the polyclonal preparations were first developed.
Recent research demonstrates that the antibody profile is greatly influenced by environmental and regional characteristics including climate, vaccination programmes, and the prevalence of pathogens in different countries and regions.116
Production of effective immunoglobulin preparations also requires a high content of monomeric immunoglobulin, important for the functionality of the purified immunoglobulin and the biological activity of the Fc-function (the part of the immunoglobulin molecule recognised by patient mononuclear cells which plays an essential function in defence against pathogens). Furthermore, distribution between the IgG1, IgG2, IgG3, and IgG4 subclasses should be similar to that found in the plasma of donors.
The first human immunoglobulin preparation to be produced on a large scale was produced using the Cohn method of cold ethanol precipitation and was called ”immune serum globulin”. During storage however, immune serum globulin solutions tended to form aggregates, generally believed to be the cause of adverse events (anti-complement activity) when this preparation was injected intravenously.117 Contaminants such as immunoglobulin M, immunoglobulin A, albumin, and plasminogen may also cause adverse events.118
Early clinical observations showed that intravenous administration of these preparations of immunoglobulin reconstituted in water, without further purification or physicochemical treatment, caused severe pyrogenic (fever-inducing) and cardiovascular reactions in many recipients.119,120
For that reason the first commercial immunoglobulin products were mostly restricted to intramuscular injections. Today, all intramuscular immunoglobulin is produced to the same standards as intravenous immunoglobulin.
Colonel Bruton introduced immunoglobulin replacement in 1952. He used a 16% immunoglobulin solution to treat a boy with agammaglobulinaemia who suffered from recurrent pneumococcal infections.121,122,123 Bruton administered the immunoglobulin monthly by the subcutaneous route and demonstrated a beneficial effect as the treatment induced measurable immunoglobulin levels and completely eliminated pneumococcal infections. These observations were rapidly confirmed and treatment with human immunoglobulin soon became the standard of care for patients with primary antibody deficiencies.124 Subcutaneous infusion, which can be performed at home, was further developed in the 2000s.
A large UK study carried out in 1956-1966 found that a high dosage of intramuscular immunoglobulin per kg body weight per week resulted in a significantly lower number of infection periods compared to a low dose. The need for another administration route became necessary as the intramuscular route was too painful and thus not suitable for high dosages.125
In 1979, a workshop sponsored by regulatory authorities on intravenous immunoglobulin (IVIg) established a consensus that IVIg products were indicated in primary deficiency patients.126
In the early 1980s, intravenous immunoglobulin manufacturing processes and formulations became available, meaning patients with primary deficiencies could be regularly treated with higher doses. In hypogammaglobulinaemia secondary to chronic leukaemia or multiple myeloma, intravenous immunoglobulin can be administered intravenously to reduce the risk of infection.
Increased demand for intravenous immunoglobulin (IVIg) was further triggered by Paul Imbach’s observation published in 1981 that the therapeutic value of IVIg could be associated with effects in addition to the passive transfer of antibodies. In the treatment of two immune deficient children suffering from serious immune thrombocytopenia (ITP), he noticed that the platelet count increased after a higher dose infusion of IVIg. This observation was confirmed by others and following the result with ITP, high doses of IVIg are administered to patients suffering from many other immune-haematological disorders, including autoimmune diseases, neurological syndromes, and other diseases of the immune system. The reasons for the successful treatment of these diseases with high doses of IVIg have not been established though a number of theories have been postulated. Although evidence-based medicine may advise against the use of a high dosage of IVIg in all these disorders, wide clinical use supported by many studies seems to show that in selected patient groups high dose IVIg treatment has a significant beneficial clinical effect.127
Thus, in addition to the production of Fraction I and cryoprecipitate (clotting factors) and Fraction V (albumin), it has become important to produce immunoglobulin at high yield for the treatment of a wide variety of primary and secondary antibody deficiencies and autoimmune diseases.
The following section sets out how manufacturing processes were refined to produce immunoglobulin that can be safely administered intravenously, and outlines a number of different product characteristics including efficacy, tolerability, and safety.
In general, Cohn’s industrial ethanol method, further refined in collaboration with Oncley, is still in use and, with some additional steps, yielded intramuscular immunoglobulin with a more satisfactory purity.128,129,130 Later modifications of Method 6 include the work by Kistler and Nitschmann in 1962 in which the number of fractions was reduced. Precipitating Fraction I+II+III as a paste in a single step increases immunoglobulin and albumin recovery and reduces costs. On the other hand, in contrast to the Oncley-Cohn ethanol fractionation process, this fraction contains more proteins.
In the Kistler and Nitschman process, the Cohn’s fractionation backbone is retained but the process is modified to adapt to the requirements of large-scale production.
Production costs are lower, as fewer fractions mean less ethanol is used (reduced from ca. 2,000 to 1,200L per 1,000 L plasma) and a smaller volume can be treated (reduced from 2.2 to 1.7 times the volume of the starting plasma).131,132,133 The reduction of the large volume of high-quality water required for fractionation is also important.
On the other hand, the immunoglobulin production yield is increased at the expense of purity. The production yield is generally between 3.5 and 4.2 g/L plasma.
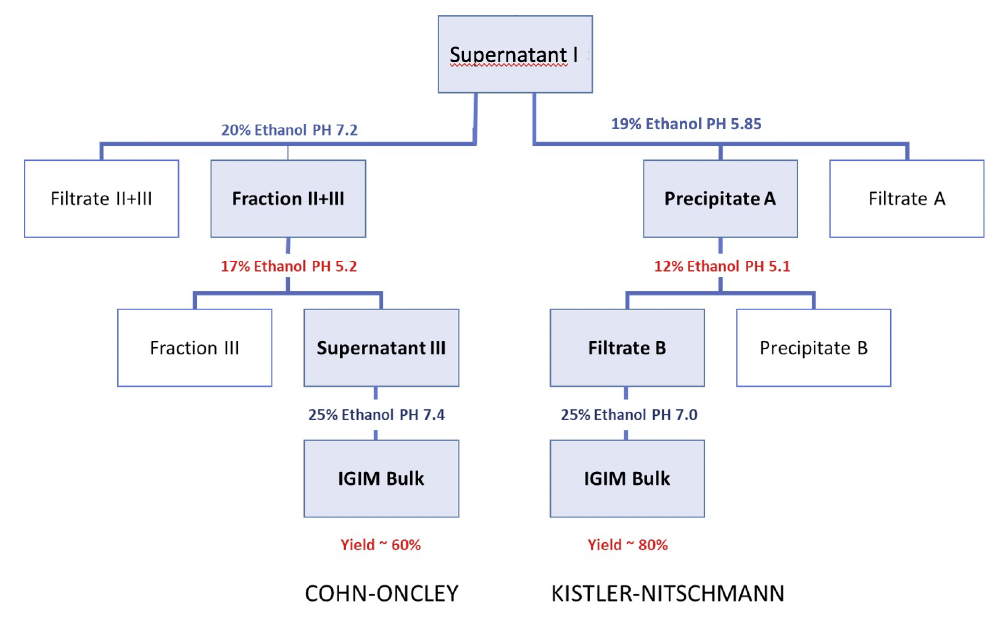
Figure 5 shows the basic modifications of Cohn fractionation in Cohn-Oncley and Kistler-Nitschmann as they are usually carried out. The starting material is Cohn Supernatant I obtained after precipitation in the presence of 8% Ethanol-pH 7.2.
Before applying either the Cohn/Oncley or Kistler-Nitschmann approach, frozen plasma is thawed at 2-4°C. The part that remains insoluble is separated and constitutes the cryoprecipitate, i.e. the starting material for Factor VIII, von Willebrand Factor, and fibrinogen.
Cohn (or Cohn/Oncley) Fraction II+III (equivalent to Precipitate A in the Kistler-Nitschmann method) is resuspended. After precipitation of Fraction III or Precipitate B (rich in lipid-bearing beta-globulin) at different ethanol concentrations, the remaining supernatant or filtrate yields, upon further ethanol precipitation, Fraction II (relatively pure, mostly immunoglobulin G) or an equivalent gamma-globulin-containing precipitate. These precipitates are then subjected to further refining processes.
The Cohn-Oncley process is still the dominant method used by most companies today with minor modifications.134 The Kistler-Nitschmann process is currently used by CSL-Behring and their licensees. The precipitate rich in immunoglobulin contains 70-89% immunoglobulin G, has 70-80% monomers and may be suitable as a preparation for intramuscular immunoglobulin injection. In these preparations, the immunoglobulin biological functions are preserved, such as its half-life in the blood circulation, complement activation in the presence of antigens and opsonising properties.135
Although immunoglobulin G produced by cold ethanol fractionation is relatively pure, it contains trace amounts of highly active contaminants. Since spontaneous complement activation by immunoglobulin G aggregates was first identified as the principal cause of adverse side effects when injected intravenously, the desire to eliminate anti-complementary activity had a significant impact on further development of intravenous immunoglobulin.136
Nearly all commercial IVIgs are produced from large pools of human plasma by first concentrating the immunoglobulin in Fraction II by cold ethanol fractionation. A series of manufacturing changes designed to reduce the incidence of side effects have been implemented (Figure 6). Contaminants have been identified, namely prekallikrein activator (which initiates production of the potent vasodilator bradykinin), activated coagulation factors, complement proteins, and immunoglobulins A and M.
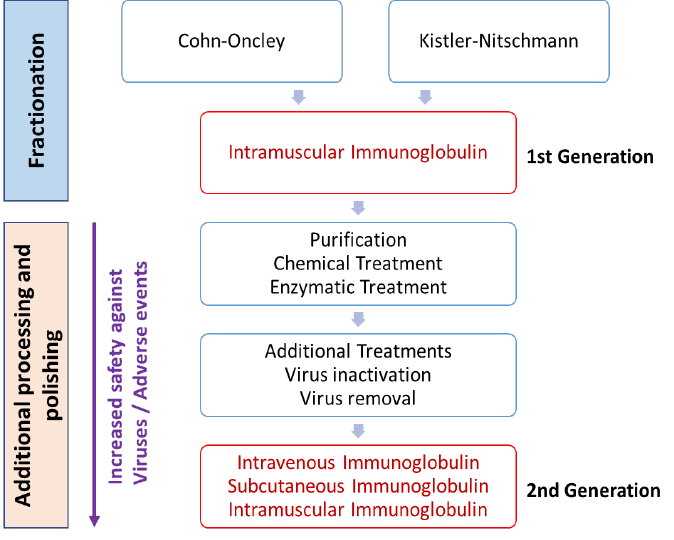
New downstream purification steps have been added, such as precipitation with caprylate (a fatty acid, also called octanoate, forming insoluble complexes with α- and β- globulins), polyethylene glycol (a non-toxic precipitant of immunoglobulin G, acting by an exclusion mechanism) or treatments at pH 4, with or without pepsin traces, or plasmin. Pepsin and plasmin are efficient specific proteases, active on aggregates.
Other methods for reducing the anti-complementary activity have been implemented, such as chemically modified immunoglobulin obtained by treatment with β-propiolactone or by sulphonation.
Unfortunately, these treatments also reduce important antibody biological activities required for clinical efficacy.138 β-propiolactone, sulphonation, and pepsin treatment induce loss of immunoglobulin subclasses. Sulfonation and pepsin treatment shorten the half-life of immunoglobulin G infusion.
The further introduction of dedicated process steps for virus inactivation or elimination will increase the number of protocols for producing safe IVIg products and their potential diversity as drugs.
Formulations and production methods for the various IVIg products vary considerably, and these differences can affect the appropriateness of selecting a particular product for some patients. For example, a final freeze-drying step may induce protein degradation.
The recognition that Cohn Fraction II contained trace amounts of highly active contaminants such as prekallikrein activator, prekallikrein, and activated coagulation factors led to the introduction of enhanced methods of fractionation. Current requirements for intravenous immunoglobulin are an intact molecule, high purity, and preferably a liquid formulation although lyophilised intravenous immunoglobulin has been available for many years.139
Besides the additional precipitation step, commercial intravenous immunoglobulins are also obtained by a combination of ethanol fractionation and new industrial chromatographic techniques developed for pharmaceutical applications.140 The chromatography process (see section 9B.7) introduced for immunoglobulins in 1972 can include several chromatography steps using different mechanisms and gel supports (ion exchange, hydrophobic, size exclusion filtration, affinity). An anion exchanger can be used in flow-through mode to capture non-immunoglobulin proteins or in batch adsorption mode. This methodology has led to the development of purified, unmodified immunoglobulin G concentrates. The removal of immunoglobulin A by chromatography decreases the risk of anaphylactic shock in patients with immunoglobulin A deficiency.
Nevertheless, with some chromatographically purified preparations, an elevated frequency of haemolytic reactions emerged after infusion into patients. Isoagglutinins, known as active thrombogenic contaminants, led the manufacturers to add additional specific immunoaffinity chromatography as a refining process (see below).141
Using chromatography technology alone allows production of high-purity immunoglobulins but does not prevent virus transmission on administration to patients.142
For further literature regarding the description and production schemes of other commercial intravenous immunoglobulin, see references 117, 129 and 135 of this report.
As already discussed, since the 1980s there have been reports of immunoglobulin-associated thromboembolic adverse events (TEEs), including myocardial infarction, ischaemic stroke, and venous thromboembolism.143 In a one-year retrospective study including 2,771 patients, new IVIg users and controls, the results led to estimating the absolute increased risk of clinical TEE attributable to IVIg at 0.7 to 0.3%. These results are in agreement with those of another retrospective study including 11,785 IVIg-treated individuals, where 122 (1%) had thrombotic events recorded on the same day as IVIg administration.144 TEEs were observed in different IVIg brands, including subcutaneous IVIgs.
Although many consider that all IVIg concentrates are similar, they vary considerably in composition as regards excipient compounds and related effects, and these differences may lead to different infusion times and have different clinical implications.145,146
Differences in biological and biochemical properties have been measured in licensed liquid IVIg preparations approved as safe and effective.147 Differences in half-life were also observed in patients after IVIg infusion. Certain patients, such as those with diabetes or those at risk of renal failure and/or heart disease might not tolerate particular IVIg formulations containing sucrose. Important variables include the concentration, volume, osmolality, sodium content, and sugar content.148,149,150,151,152
Adverse events occur in less than 5% of patients.153 As previously mentioned, a potentially elevated TEE frequency has been found for different IVIg products, including subcutaneous ones. This may be related to product manufacturing processes.154
This data requires explanation, as recent results suggest new applications of IVIg products in the treatment of highly HLA-sensitised (Human Leukocyte Antigen) patients who are awaiting transplantation, for the management of viral (e.g. parvovirus) infections, and for the treatment of antibody-mediated rejection (see Table 1).155,156
Passive immunisation with immunoglobulin is recommended, for instance, for prophylaxis against hepatitis A and against infection by its causative agent, the hepatitis A virus. Because anti-hepatitis A antibodies are declining in the donor population and hence, in immunoglobulin products, differences can be observed between different commercial immunoglobulin products.157
Regulatory authorities therefore require clinical trials with more standardised protocols and standardised tests to quantify specific antibodies, and pharmacokinetic studies providing more meaningful information with respect to appropriate IVIg doses, notably to prevent infection and TEEs. The US Food and Drug Administration (FDA) has also mandated that IVIg products include a prominent boxed warning about TEEs.158,159
The following section sets out how manufacturing processes have improved to produce new PDMPs, focusing in particular on chromatography.
Since Cohn ethanol fractionation was established, progress has been made with the introduction of new precipitation techniques such as glycine, polyethylene glycol, caprylate, rivanol, and ammonium sulphate precipitation. From the 1970s to the present, industrial development has been continuous with the emergence of new biotechnological and pharmaceutical processes. This progress has been bolstered by advances in biotechnology, notably in pharmaceuticals and food processing, with the introduction of chromatography (explained below), centrifugation, depth and sterile filtration, ultrafiltration, and lyophilisation. Press filters used in place of centrifugation have made larger-scale production possible.
Plasma fractionators have also introduced improvements to produce PDMPs with a higher yield and better therapeutic properties.
The demand for clotting factors, immunoglobulin, and albumin is increased, pushing both commercial and institutional fractionators to expand their facilities, increase their production capacities, and meet patients’ needs.160,161,162 As a result of larger-scale industrial production, a plasma pool of up to 1,000 litres could be processed as a single batch, expediting plasma processing and the manufacture of plasma-derived products.163
The global volume of plasma collected has increased continually: from 10 million litres in 1982 to 17 million in 1995, 22.5 million in 2007, and 40-45 million in 2018.164,165,166,167,168
The introduction of chromatography (a laboratory technique known since the 1950s) in the late 1970s to early 1980s was a major technical innovation. Chromatography is a technique in which the mixture to be separated is dissolved in a fluid (gas or solvent) called the mobile phase, which carries it through a system (a column, a capillary tube, a plate, or a sheet) on which a material called the stationary phase is fixed. The different molecules stay longer or shorter on the stationary phase, depending on their interactions with its surface sites, causing them to separate.
In contrast to Cohn fractionation and similar to the case of immunoglobulin, chromatography is recognised as a purification technique capable of obtaining high purity with high yields. It is also easily automated and more versatile than precipitation.169
The advantages of chromatography over other fractionation methods include its easy integration with the routine Cohn method, its relative rapidity, and the fact that it doesn’t require much space in the manufacturing plant.170,171 The Cohn ethanol fractionation process requires more space and high-capacity tanks (today, 3,000 to 50,000 litres) with efficient cooling systems to reach temperatures below -8°C inside the product. This requires significant cooling capacity and anti-explosive materials. Furthermore, the ethanol-rich waste produced (which can reach 40%) requires specific handling.
New materials have emerged as stationary phases for chromatography which bind the protein of interest strongly but reversibly and with high specificity and selectivity. In gel filtration chromatography, which separates macromolecules according to size (large molecules flow faster through the stationary phase), non-specific binding is preferable. Different chromatographic separations are available for ion exchange and affinity chromatography, and these can be used in batch mode (the proteins to be separated are mixed with the stationary phase) or in flow-through mode (under anion- or cation-exchange conditions, for example). The corresponding resins must have a reproducible performance and be suitable for sanitisation in place or even cleaning in place to avoid endotoxins and sticking impurities such as viruses liable to remain present in the next purification process. As such, chromatography is a capable industrial-scale technology with few and simple scaling-up parameters and may be a potent virus removal step. Its virus removal capacity depends on the nature of the virus, the viral load, and the conditions used.
Since the 1970s, new purification processes have emerged with or without chromatography, starting from the cryoprecipitate or from the cryosupernatant produced before the first ethanol precipitation step. This has expanded the number of clotting concentrates to include FVIII, von Willebrand Factor, FIX, Prothrombin concentrate or PPSB, and fibrinogen (Figures 4a and 4b). It must be stressed that patients requiring PDMPs may be exposed for their whole life and are therefore susceptible to adverse and undesirable effects. New, improved products for treatment should be carefully analysed and monitored through pharmacovigilance. In parallel, research on plasma proteins has increased knowledge and led to improved laboratory analysis techniques.
Fractionators have retained Cohn fractionation for parts of the production process, as this strategy offers industrial advantages: lower development costs and lower production costs, including reduced use of human resources and less costly process quality control. Another noteworthy advantage is that PDMPs are already on the market and have complete drug approval files in most countries.
The different fractions are thus used as starting materials for downstream processes, yielding new products. Then, common validation steps can be performed (Figures 4a and 4b).
A variety of new PDMPs have been developed and have entered the therapeutic panel of PDMPS which are concentrated plasma protein products that have therapeutic value.172,173,174,175 They include antithrombin III and thrombin (using immobilised heparin on gel), α-1 proteinase inhibitor (precipitation followed by ion-exchange chromatography or ion exchange followed by gel filtration according to the manufacturer), and C1-esterase inhibitor concentrate (chromatography).
Most fractionators maintain cold ethanol fractionation for purifying albumin.
Two fractionators, BPL (Bio Products Laboratory Ltd) and CSL Plasma, have introduced one or more chromatography steps for this purpose.176 The flow chart of BPL albumin (“Zenalb”) production includes the ethanol fractionation steps plus a final anion-exchange step. The CSL process applies three chromatography steps, starting with Supernatant I obtained by cold ethanol fractionation. BPL and CSL fractionators claim a higher purity for the albumin produced by these methods.
The following section provides information on viruses which have been transmitted through plasma products, focusing particularly on transmission through clotting factor concentrates, polyvalent immunoglobulin and anti D immunoglobulin.
Although major progress has been made in transfusion medicine to improve blood safety with regard to blood borne viruses, complete elimination of the risk of transfusion reactions and transfusion-transmitted infections has not been achieved. Immune reactions due to alloantibodies and sensitisation to HLA-antigens also constitute an important cause of side effects related to transfusion. The data on adverse effects are incomplete since a number of side effects can only be observed after months or years.
Blood and blood products are known to transmit a variety of infectious diseases. Since the 1970s, transmission of several pathogens via plasma-derived medicinal products has been reported (Table 3).
|
Pathogen |
Characteristic |
Disease |
Period |
|---|---|---|---|
|
Plasmodium |
protozoa |
malaria |
1911 |
|
Treponema pallidum |
bacteria |
syphilis |
1940s |
|
Serum hepatitis virus (early findings ) |
virus |
hepatitis |
1933 – 42 |
|
Hepatitis B virus identification (4) |
enveloped virus (1) |
hepatitis |
1960s |
|
Babesiosa |
protozoa |
babesiosis |
late 1970 |
|
Hepatitis non A-non B virus later identified as Hepatitis C virus |
enveloped virus |
hepatitis |
1970s |
|
HIV 1 (HTLV-III) & HIV 2 |
enveloped virus |
Acquired Immune Deficiency Syndrome (AIDS) |
1982 |
|
Hepatitis A virus |
non-enveloped (2) |
hepatitis |
1940s |
|
Parvovirus B19 |
non-enveloped |
5th disease |
1990 |
|
West Nile virus |
enveloped |
WN disease |
1999 |
|
vCJD (3) |
infectious prion |
variant Creutzfeldt Jakob disease |
1996 |
|
Dengue virus |
enveloped |
dengue |
2002 |
|
Chikungunya virus |
enveloped |
chikungunya |
2000 |
|
Hepatitis E virus |
non-enveloped |
Hepatitis |
2004 |
|
Zika virus |
enveloped |
Zika disease |
2007 |
|
Trypanosoma cruzi |
protozoa |
Chagas disease |
2011 |
(1) Virus with an outermost layer or coat made of lipids and proteins. (2) Virus without a coat. (3) vCJD = variant “Creutzfeldt-Jakob disease”. It is a transmissible spongiform encephalopathy with infectious prion-modified natural protein (see text section 9B.8.v). (4) The existence of hepatitis transmitted by human serum was established in the late 1930s – 1940s. The term hepatitis B was introduced in 1947. The virus was identified by Blumberg in the 1960s.177,178
The occurrence of viral hepatitis A, hepatitis B, hepatitis C and hepatitis E, human immunodeficiency virus (HIV) – the causative agent of acquired immunodeficiency syndrome (AIDS), human T-cell lymphotropic viruses HTLV I and II, the parvoviruses, cytomegalovirus and in recent years West Nile virus, and Zika virus following transfusion, underline the importance of being aware of protecting against the risks of transmission of infections through blood and plasma products. The approaches to improve the safety of blood and plasma products are based on information to potential donors, careful selection of donors, screening and testing of donated units for known infective agents and the use of validated fractionation methods, which include specific steps designed to remove or inactivate viruses. The virucidal methods employed are reported in response to Question 10.179
In the early 1980s the first evidence for transmission of AIDS by clotting factor concentrates prepared from pooled plasma became apparent. Subsequently it was demonstrated that Human Immunodeficiency Virus (HIV) present in donor plasma when transferred to recipients of blood and blood/plasma products may cause post-transfusion AIDS. During subsequent years, more extensive donor screening procedures and the introduction of viral inactivation steps such as dry-heat treatment and pasteurisation have significantly reduced the risk of transmitting several viral diseases (see Questions 10 & 11).180
However, when assays became available for the detection of hepatitis C virus, which accounts for 90-95% of cases of post-transfusion hepatitis, it was shown that not all viral inactivation steps were sufficient to prevent transmission. One of the consequences was that cryoprecipitate not treated by efficient virus inactivation/removal techniques was no longer recommended for the treatment of people with haemophilia.181
Transmission of hepatitis C remains a problem in many areas of the world where haemophilia is treated with cryoprecipitate and 80% of the individuals using cryoprecipitate are estimated to be infected with hepatitis C.182
Viral inactivation through mixtures of solvent and detergent and chromatographic purification procedures has proven to be effective in the elimination of hepatitis B and C virus.
Despite the aforementioned viral inactivation steps, an isolated case of transmission of hepatitis C virus through pasteurised Factor VIII was reported in a young patient with haemophilia, but this case was questioned because hepatitis C is not exclusively transmitted by blood and blood products.183,184
The case reported in 1992 concerned a haemophilic patient of 7 years of age who had received substitution of Factor VIII (Haemate P, Behringwerke), and who had never undergone surgery or received other blood products. Routine testing for HCV-Ab, confirmed with 4-RIBA and HCV-PCR and elevated ALT revealed HCV positivity. During the year before seroconversion this patient had received Haemate P from five different batches. These batches were analysed for HCV-RNA with PCR, using primers from the 5’ non-coding region, and two batches were positive. PCR had been routine at the laboratory for more than 6 months and no contamination was observed. After thorough investigation, transfusion with any other plasma products could be excluded. Although this isolated case seemed to show that pasteurised (60°C for 10 h in solution) Factor VIII may transmit HCV, no other case of HCV transmission had been definitively connected with Haemate P over 10 years. This case was questioned because 7 patients of the same centre who received Haemate P of at least one of the two positive batches half a year before or more remained anti-HCV positive. Moreover, isolated observations of HCV infection in haemophilic patients are to be expected since the infection is not exclusively associated with transmission of blood and blood derivatives.
Several cases of hepatitis A were observed in 83 haemophilia A patients in Italy, Belgium, Ireland and Germany after treatment with a high purity Factor VIII concentrate. The viral inactivation step during the manufacturing process was solvent detergent, which is known not to inactivate non-enveloped viruses such as HAV. In some of the batches of the implicated concentrate, HAV RNA was demonstrated. However, HAV could not be detected in the process water used during the manufacturing process.185,186
Parvovirus B19 infections are common and mostly without clinical significance except for pregnant women (prevalence 1/40,000) with some significant events reported in 10% of cases187,188,189 and also people with weakened immune systems.190
Clinical experience with IVIg products has been extensive with millions of doses administered in the past 20-30 years.191
There have been a few exceptional cases of HBV transmission in the past through intramuscular immunoglobulins made from plasma collected before third-generation hepatitis B surface antigen (HBsAg) testing was performed.192
It was thought that the cold liquid ethanol fractionation procedure and purification methods such as DEAE Sephadex and adsorption chromatography decreased the risk of virus transmission. But the identification of the causative virus (or viruses) in patients by serologic techniques was difficult because most of the patients involved had severe immunodeficiency disorders and were unable to mount an antibody response.193
In the 1980s, Non-A and Non-B virus (NANBH) was reported to be transmitted in immune deficient patients receiving IVIg preparations. Explanations tended to focus on the experimental IVIg production process performed in Sweden, the UK and the US with regards to the variation of the original Cohn fractionation (Fraction I was not removed), the absence of freeze drying in the presence of alcohol, and failures to comply with Good Manufacturing Practice (GMP) rules.
Other reports of NANBH transmission involved IVIg preparations in which an adsorption chromatography step was used, even if one preparation was treated by pH4+ pepsin (a single batch). In 1994, the FDA was notified of 112 possible cases of HCV associated with Gammagard (Baxter) over 1993-1994. More than 60% of people treated with Gammagard were infected with HCV.194 Gammagard was produced from very large pools and cold ethanol fractionation was used followed by chromatography. It was licenced in the US in 1986 and had a good safety record.195 In 1991, anti-HCV antibody testing was introduced to increase plasma safety with all positive donations discarded.
The introduction of serologic tests for HCV in 1991 created a problem for the immunoglobulin manufacturers. Although many batches of immunoglobulin concentrates, made before the testing, were found with detectable viral HCV RNA in later studies, no virus transmission was reported. These former immunoglobulin preparations were made from large pools of plasma containing HCV and in most cases no deliberate virucidal step was used in their manufacture process. It was demonstrated that small amounts of HCV particles contaminating the plasma pools were neutralised by the presence of specific anti-HCV antibodies, and forming complexes with the virus contributed to some extent in this way to HCV infectivity removal by the Cohn fractionation process.
There was some evidence, based on retrospective studies and a few experimental studies on chimpanzees, that the non-tested immunoglobulin preparations may protect infused patients from further infection.
Using first-generation (single antigen) screening tests to detect anti-HCV in donations allowed many positive donations (containing the complex anti-HCV/HCV) to be discarded before pooling, but left some unbound viruses in the future pool. This was overcome by the use of second-generation anti-HCV testing and the introduction of specific virus inactivation steps in the immunoglobulin production process.196,197,198,199,200,201
As mentioned, between October 1993 and April 1994, a further intravenous immunoglobulin product (Gammagard) was implicated in the transmission of HCV. Infection was associated with higher quantities of HCV RNA in the IVIg product produced from second-generation anti-HCV-screened plasma.202,203
Transmission of hepatitis C has been suspected in transplant patients who received cytomegalovirus immunoglobulin. HCV antibodies were transferred passively by CMV immunoglobulin. When the treatment was stopped, anti-HCV could not be detected in the blood samples after 12 weeks.204
Manufacturers of human immunoglobulin introduced additional inactivation steps such as pasteurisation (10-hour heat treatment in the liquid state), S-sulfonation, B-propiolactone, and treatment with TNBP (tri(n-butyl) phosphate) and sodium cholate to further improve the safety of human immunoglobulin.
Human anti-D immunoglobulin is a liquid or freeze-dried preparation containing immunoglobulin, mainly immunoglobulin G. The products contain 100-180 grams of protein with a high titre of Rh-D antibodies. The protein fraction consists of at least 90% immunoglobulin G (IgG), small amounts of immunoglobulin A (IgA) and immunoglobulin M (IgM) and traces of other plasma proteins. The product supplied is sterile and pyrogen-free (i.e. not causing fever).
The preparation is intended for intramuscular or intravenous use. It contains specific antibodies against erythrocyte D-antigen and may also contain small quantities of other blood group antibodies.
The product is preferably obtained from plasma of donors with a sufficient titre of previously acquired anti-D antibodies. Where necessary, in order to ensure an adequate supply of human anti-D immunoglobulin, it is obtained from plasma derived from donors immunised with D-positive erythrocytes that are compatible in relevant blood group systems of the donor in order to avoid the formation of undesirable blood group antibodies.
The requirements for intravenous products, which are additional to those for intramuscular preparations, include tests and limits for anti-complementary activity and for anti-A and anti-B haemagglutinins.
The products are obtained from the pooled plasma of donors with sufficient titres of anti-D antibodies. Screening includes tests for the absence of markers for hepatitis B (HBV), hepatitis C Virus (HCV) and Human Immunodeficiency Virus (HIV) and an upper limit of 10E4 IU/ml is applied for B19 virus DNA. Manufacture includes validated purification and virus inactivation/removal steps.205,206
In the UK the plasma used in production by BPL since the early 1960s has been consistently provided by the National Blood Transfusion Service from voluntary, non-remunerated blood donors, who have a suitable high titre of antibodies to the Rh-D antigen, and, over the period in question, increasingly effective screening systems have been introduced to exclude hepatitis B Virus and Human Immunodeficiency Viruses.
The current procedures for selection of donors, screening of plasma, the specification of immune plasma and procedures for its incorporation into fractionation are all included in the manufacturing protocol. With the development of new blood collection systems, the immune plasma used in the process has changed from solely fresh frozen plasma recovered from whole blood donations, to plasma collected either from whole blood donations or, increasingly, through automated apheresis procedures. The process of purification involving ethanol fractionation has remained essentially the same since 1965; the process changes that have been introduced relate to the scale of the new manufacturing unit.207
Primary immunisation and boosting of volunteer Rhesus-D negative blood donors with Rhesus-D positive red cells has been undertaken in the UK since around 1970. Since no alternative to human polyclonal anti-D immunoglobulin is available for prophylaxis and given the shortage of naturally immunised donors, primary immunisation and boosting of volunteers is considered fully justified. The procedures used have been carefully designed, are kept under regular review and are updated to minimise any potential or theoretical risks.208
Following treatment in 1979 with contaminated anti-D immunoglobulin, several outbreaks of hepatitis Non-A Non-B occurred in women in various centres in the former German Democratic Republic. The immunoglobulin had been prepared from small pools of ten donors by DEAE-Sephadex A 50 chromatography followed by precipitation with 25% ethanol (final concentration) and sterile filtration. Infection with hepatotropic viruses (HAV, HBV, Cytomegalovirus (CMV), Epstein Barr virus (EBV)) and metabolic, allergic and autoimmune liver diseases were excluded in recipients of contaminated immunoglobulin. The retrospective investigation of the outbreak revealed two plasma donors who developed acute hepatitis following the administration of erythrocytes from a rhesus-positive donor with a hepatitis infection which was not clinically apparent. The occurrence of antibodies to hepatitis C virus (HCV) was investigated in 81 patients who developed hepatitis Non-A Non-B after parenteral administration of contaminated immunoglobulin to prevent Rh sensitisation. 90% of patients infected by a common source became anti-HCV positive.209,210
1,037 patients were contaminated by many batches of anti-D immunoglobulin produced by ion-exchange chromatography with no cold ethanol fractionation step used before chromatography.211,212
It is likely that the ethanol precipitation after DEAE-Sephadex chromatography was insufficiently effective/virucidal to ensure the safety of the anti-D immunoglobulin preparation.213
In 1977 in Ireland, 704 people received hepatitis C infected anti-D immunoglobulin and in 1991, 72 people. In the anti-D recipient contact programme, 11 people were identified, in the targeted lookback programme, 61 recipients of transfusion from donors were directly infected by anti-D immunoglobulin, and 1 person was infected in the optional screening programme. All cases could be linked to an anti-D immunoglobulin product manufactured by ion exchange chromatography together with an ethanol precipitation step, which was considered beneficial in helping to inactivate any hepatitis viral material that might be contained in the plasma being fractionated.214 The source material for manufacturing five batches of anti-D immunoglobulin included plasma from a patient who underwent a course of plasma exchange to try and prevent the Rh haemolytic disease. This patient later developed hepatitis.
In Ireland between January 1991 and January 1994, 46 batches of anti-D immunoglobulin were produced including 10 donations in 1989 from a donor who tested negative for hepatitis C. The infection probably arose from a failure to detect a donor carrying the hepatitis C virus before the introduction of hepatitis C screening in October 1991.215
Clearly, the new chromatographic method (DEAE-Sephadex A 50) developed in East Germany, and used in Ireland and Germany in order to maximise the yield of anti-D immunoglobulin production was a total change in the manufacture of immunoglobulin. That chromatography process followed by an ethanol precipitation (25%) appeared to have too little capacity to remove the HCV viral load present in the starting plasma pools during the immunoglobulin production.216
In 1998, the British Government urgently advised Rhesus negative mothers who had recently delivered a Rhesus positive child to avoid injection with Rh-D immunoglobulin because of the risk of contamination with a new variant of Creutzfeldt-Jakob Disease. The background to this advice was that the available stocks of Rh-D Immunoglobulin were derived from plasma of British blood donors who theoretically could be contaminated with the human variant of Bovine Spongiform Encephalitis (BSE) (see also section 9B.8.v).217
Variant Creutzfeldt-Jakob Disease (vCJD) is a relatively new, rare and untreatable neurodegenerative zoonotic disease, classified as a transmissible spongiform encephalopathy (TSE) or a prion disease. vCJD was first identified in March 1996 in the UK when 10 cases of a new disease with neurologic symptoms were reported in UK patients of an unusual young age (median 29 years). Soon thereafter, it was associated with the bovine spongiform encephalopathy (BSE) disease in the UK, referred to as “mad cow disease”, a novel disease affecting cattle.
In prion diseases, abnormal disease-specific protein aggregates accumulation is found in the brain. A unique feature of the prion disease is that it may also be acquired and transmit infection between individuals of the same species and in some instances between different species.218,219
After the outbreak of Bovine Spongiform Encephalitis and variant Creutzfeldt-Jakob Disease in the UK, the UK Department of Health and UK Blood Services implemented several vCJD risk-reduction measures for blood, blood products and blood components. Between 1999 and 2021, plasma for the manufacture of fractionated plasma products such as immunoglobulins was obtained from sources outside the UK. The main source of plasma for these products was the US.
As a precautionary measure, removal of leukocytes (white blood cells) – leucodepletion – was introduced for all blood products in several European countries because the prion protein responsible for vCJD probably binds to leukocytes. In a number of countries, donors who lived in the UK for a period of six or more months between 1980 and 1996 are excluded from blood and plasma donation.
Currently, 230 vCJD cases have been reported worldwide in 12 countries including Taiwan (1), Japan (1), Saudi Arabia (1), Canada (2) and US (4). Since 1995 and as at 30 July 2021, 223 vCJD cases (159 confirmed and 64 probable cases) have been reported from seven EU/EEA countries, with the highest number of cases reported in the UK (178) and in France (27). The last known UK case of vCJD was reported in 2016. The majority of vCJD cases were primary transmissions associated with dietary exposure to BSE (Bovine Spongiform Encephalopathy) agents in beef. Secondary transmission through exposure to transfusion of plasma derived medicinal products with the presence of transmissible proteinaceous infectious particles – called prions (PrPsc)- have only been reported in the UK. A total of five people died following transmission of vCJD from blood donors who later developed vCJD; two of them died from another cause with no symptoms of brain disease but were found to have evidence of vCJD in their spleen, which could represent a pre-clinical form of the disease. All infected recipients had received red cells that had not been leucodepleted.220,221
Due to limitations in the supply of immunoglobulin products caused by the need to import plasma in the UK to supply patients with the highest clinical need, the pressure on supply due to the increased need for blood plasma in the development of new Covid-19 medicines and the falling numbers of donations, the United Kingdom lifted its ban on the use of UK-sourced plasma to produce immunoglobulin products in February 2021. The UK assessed the variant Creutzfeldt-Jakob disease (vCJD) risk for immunoglobulin products manufactured from UK plasma as very low and acceptable in the context of overall vCJD risk in the general population, taking into consideration a risk-benefit analysis.222
This opinion was however not shared by the European Centre for Disease Prevention and Control that stated that the absence of a reliable diagnostic blood test makes it difficult to assess the residual risk for transmission of vCJD infection through blood components and PMPDs obtained from UK-sourced blood and plasma donations with any degree of confidence.223
The following section starts with an outline of the work of Dr Judith Pool and colleagues on cryoprecipitate. It then briefly covers the early development of Factor VIII concentrates and the work of the Oxford Group on Factor IX concentrates.
The history of concentrated Factor VIII began in the early 1940s when Edwin J. Cohn pioneered fractionation of plasma with various concentrations of ethanol. The first fraction so obtained is called “Fraction I”.224
This ‘Fraction I’ contained fibrinogen and Factor VIII, although the methods of assay had not been developed for this fraction, nor had von Willebrand Factor been identified (this happened in 1971). The value of Fraction I in treating haemophilia was demonstrated early and modest amounts were used in developed countries throughout the 1950s and 1960s, but sterile production required a large, well equipped laboratory.225,226
In Nancy, France in 1955, Dr Emile Rémigy published his thesis “Essais de traitement par plasma concentré” (Trials of treatment with cryo-concentrated plasma) in which he described the in vivo correction of the prothrombin consumption time in three severe haemophilia A patients by injecting intravenously 250 ml of cryo-concentrated plasma (cryoprecipitate). His thesis was based on the procedure of cryo-concentration of plasma and proved interesting in the initial treatment of haemophilia due to the product’s reduced volume and enlarged concentration of proteins. The presence of fibrinogen in an insoluble cryoprotein fraction obtained from frozen plasma thawed at 4°C was published by Ware and colleagues in 1947. In 1948, Morrison and colleagues described the precipitation at a low temperature of a fibrinogen-rich globulin preparation from Fraction I of human plasma as a method for separation of purified fibrinogen.227
In 1956, the first concentrate (fraction I-O) was produced starting from Fraction I and used in Sweden under the commercial name AHF-Kabi. It contained fibrinogen, Factor VIII, native von Willebrand Factor and Factor XIII.
It is purified 20-40 times when compared with fresh plasma.228 A commercial version of Fraction I became available in the US as a concentrate of fibrinogen rich in Factor VIII; in a one-stage assay, the ratio of Factor VIII to total protein was seven times that of natural plasma.229
At that time, blood banks were separating and freezing plasma from whole blood for local use. However, the haemostatic efficacy of whole plasma was suboptimal because only a limited volume could be infused at one time.
In 1959, Dr Judith Pool at Stanford University (USA) developed the ‘two-stage’ assay for Factor VIII and she used that method to assay Factor VIII in plasma fractions. In the same year Dr Pool and her colleagues discovered that the residue left by Cohn fractionation, which remained at low temperature after drawing off the liquid produced in the thawing process, contained a high concentration of fibrinogen and anti-haemophilic activity (Factor VIII). In 1964, Dr Pool described a method of producing these concentrated factors from plasma by freezing which was quite independent of the need for Cohn fractionation. Their starting material was pooled plasma, frozen in large containers, which they thawed gradually. They knew that the potency of Factor VIII in plasma dwindled at room temperature, so they were careful not to thaw the plasma completely. They found very little Factor VIII in the thawed supernatant. They then tested the small amount of unthawed, fibrous-looking paste at the bottom of the containers and found Factor VIII. The residue also contained von Willebrand’s Factor and other proteins, including fibronectin.230
The process devised by Dr Pool and her colleagues separated plasma from the red cells in whole blood donations by centrifugation at 4˚C as soon as possible after collection in the normal way, using standard compartmented plastic bags. The tubing connecting the compartments of the bag was clamped. The satellite bag containing the plasma was fast frozen. The whole bag was then refrigerated for cold thawing of the frozen plasma. When thawed to only a few degrees above zero, and typically to 4˚C, fibrinogen precipitated as a ‘sludge’ containing much of the Factor VIII content of the plasma. The temporary clamps were removed and the supernatant plasma was allowed to return to the red cell bag. The bags were separated and the cryoprecipitate frozen for storage pending use. The process did not produce mutually exclusive components. Red cells used in clinical practice contained very small amounts of plasma, platelets were suspended in plasma and each fraction had the potential to transmit infection. However, the Pool and Shannon method produced a cryoprecipitate that was high in Factor VIII content and was stable and soluble.231
This provided a relatively purified form of Factor VIII for haemophilia therapy. Attempts had been made to isolate Factor VIII for clinical use in the treatment of haemophilia in the 1930s. But only now was there a relatively straightforward and effective procedure.
Cryoprecipitation of Factor VIII from single units of fresh-frozen plasma was viewed as a simple, practical procedure that could be carried out by any blood bank. Blood transfusion centres in many countries used it. The single cryoprecipitate units might thereafter be pooled for further processing, but typically were used in multiples to make up a dose for haemophilia A therapy. The product had lower coagulant activity than the material produced by Cohn Fraction I. But it was inexpensive to produce and processing extra plasma could make up for the deficiency in coagulant activity. However, the method depended on prompt processing after collection when coagulant activity was high, and new technology was required to process plasma on a large scale.
When cryoprecipitate from 10–15 individual plasma donations was combined and given to the patient it was possible to raise the Factor VIII level sufficiently to stop haemorrhage. During the late 1960s this treatment became available to haemophilia patients at hospitals on an outpatient basis. This was a major therapeutic advance in the treatment of haemophilia A.
As cryoprecipitate does not contain very much Factor IX it was unsuitable for the treatment of haemophilia B.232
The manufacturing process has changed little since first described by Pool. Cryoprecipitate is prepared by thawing 1 unit of frozen plasma or fresh frozen plasma at 1°C to 6°C. After the product is thawed and centrifuged at 5,000 × g for 6 minutes, the supernatant is removed leaving the cold insoluble precipitate plus 5 to 15 mL of plasma in the original bag. This residual material is refrozen within 1 hour of thawing and stored at −18°C or colder. The resulting product has a shelf-life of 12 months. Methods of manufacturing may vary slightly in different jurisdictions. Cryoprecipitate cannot be reliably virus-inactivated. The use of this form of replacement therapy was limited and a risk of transfusion-transmitted infections could not be ruled out. However, due to the very small number of donations which form the basis for the production of cryoprecipitate, the number of transmissions was very low compared to the Factor VIII products. Being a blood component, adverse events related to cryoprecipitate are haemolytic transfusion reactions, febrile non-hemolytic reactions, allergic reactions ranging from hives to anaphylaxis, septic reactions, Transfusion Related Acute Lung Injury (TRALI), circulatory overload, and transfusion associated graft versus host disease. Currently cryoprecipitate is used as fibrinogen replacement, Factor XIII replacement, Factor VIII replacement, and von Willebrand Factor replacement. Per unit, cryoprecipitate has the same risk of viral transmission as a unit of red blood cells or Fresh Frozen Plasma; however, given that an adult dose of the product is 8 to 16 units, this product has a greater risk of transmission per dose. In haemovigilance reports, adverse event rates for cryoprecipitate are rarely reported. In a report from the haemovigilance program in Quebec, Canada, the rate of all adverse events from cryoprecipitate in 2001 was 6 per 10,000.233
The industrial manufacturing of plasma-derived enriched Factor VIII and Factor IX increased the availability of the supplies. In these first years, the life expectancy of haemophilia patients increased dramatically.234 In the late 1950s, half of the patients with haemophilia would die by the age of 19 years, whereas the median life expectancy reached approximately 50 years in the western world in the early 1980s. More specifically, for the time period that cryoprecipitate was the main treatment for people with haemophilia, a life expectancy of 57 years from birth was reported for the 1960s and 1970s in Sweden, 63 years from birth from 1973 to 1985 in the Netherlands, and 61 years from age 1 year from 1971 to 1980 in the United States.235
The industrial manufacturing of plasma-derived enriched Factor VIII and Factor IX as well as cryoprecipitate broadened the availability of the supplies. Since the introduction of anti-haemophilic factor (AHF or Factor VIII:C or Factor VIII), the amount of FVIII used per year has increased steadily in the Western world. About 80 million units were produced in 1971, 400 million in 1976, 687 million in 1979 and approximately one billion 330 million in 1984.236 The advantage of concentrates is that it is easy to maintain a high Factor VIII level with small volumes of concentrates. The disadvantage is that concentrates may be made from large pools of plasma (about 5,000 donors in 1975) and the risk that one or more donors may have had a subclinical infection increased.
In order to manufacture PDMPs in an efficient production process, the size of the production plasma pool is important. The production of polyvalent immunoglobulin requires a plasma pool of at least 1,000 donations. As previously mentioned, the disadvantage of a large plasma pool is however that one infected donation will contaminate the complete plasma pool.
Starting from cryoprecipitate, purified and highly concentrated commercial freeze dried (lyophilised) FVIIII preparation was developed using various technologies.
The first Factor VIII concentrate developed in the early 1960s was an experimental freeze dried product from Cutter Laboratories (US). However, the yield from plasma was very low, and Cutter could not produce much of this material.237
Factor VIII concentrate could also be prepared from animal plasma. Bidwell prepared Factor VIII concentrates from bovine and porcine plasma, which were purified 100-4,000 times. These products were used for the treatment of haemophilia. The haemostatic effect was good on a short-term basis, but patients developed antibodies and sometimes manifested severe allergic reactions.238,239
Developments since the 1960s have led to the availability of more and more concentrated Factor VIII products, purified by different methods including multiple precipitations and different types of chromatography (ion-exchange chromatography, binding to heparin-ligand chromatography, or immunoaffinity chromatography using monoclonal antibodies).240 The resultant concentrates might differ in certain elements. For example, the content of von Willebrand Factor varies substantially from one product to another.
To minimise the viral infective risk, manufacturers adopted methods of viral inactivation in the late 1980s and in the 1990s: solvent-detergent, dry heat, pasteurisation and vapour (see Questions 10 & 11).
Treatment of patients with haemophilia B was initially with fresh-frozen plasma. Until 1967 that was the only treatment available for correcting deficiencies in coagulation Factors II, VII, IX and X. In 1959 Professor Jean-Pierre Soulier working at the National Blood Transfusion Centre in Paris discovered prothrombin complex concentrate (PPSB) which contained prothrombin, Factor VII, Factor X as well as Factor IX, a plasma derivative for the treatment of haemophilia B patients. The demand for PPSB, which proved to have wide-ranging application, prompted research into new methods of recovering Factor IX from the normal citrated plasma used in Cohn fractionation. The Oxford Group began their Factor IX fractionation programme early in 1959. Their research led to development of a Factor IX product, based on ion exchange purification. In a similar fashion to Factor VIII, it had as its aim the production of a finished product with a specified amount of Factor IX activity per vial which would be suitable for home therapy and which would comply with the specifications of the British Pharmacopoeia (the UK standards for medicinal products).241
In England at that time large-scale fractionation of plasma was carried out at the Blood Products Laboratory at the Lister Institute at Elstree using the process developed by Kekwick & Mackay.242
This yielded as a by-product a yellowish-green, greasy final residue, known as G2, which was rich in Factor IX. At Oxford it was decided that it would make sound economic sense to use this fraction as a starting material. In March 1959, they started with the development of a reliable Factor IX assay, which could be used in plasma as well as in concentrate. By the end of January 1960, an assay was available which performed satisfactorily. The production of Factor IX was not without problems, mainly related to the method of sterilisation. Another cause of concern was the possibility that large amounts (2,000-4,000 units/ml) of prothrombin present in the concentrate might activate to thrombin, with disastrous consequences in the patient. While the patient had a disease with a (severe) bleeding tendency, the adverse event following the high dose treatment with PPSB was clotting resulting in thrombosis, stroke or heart attack which ended in some cases in death. To counter this possibility, heparin was added to the product and the citrate content was maintained by dialysing against citrate/saline.
Between 1960 and 1995, many changes were made in the fractionation process used in the manufacture of Factor IX. The early preparations made in Paris and Oxford and by several commercial manufacturers were prothrombin complex concentrates. Purified plasma-derived Factor IX concentrates were obtained by anionic chromatography or by immunoaffinity or by both types of purification. The viral inactivation technologies introduced in their production were solvent-detergent, dry heat, detergent, vapour, and nanofiltration or thiocyanate.243 The great majority of manufacturers used two of these virus inactivation technologies in their processes.244
In 1965, 1,000 litres of plasma was the volume of plasma that was fractionated weekly in contrast to 3,000 litres of plasma per week in 1972. By this time, furthermore, plasma pools of 1,000 litres and more could be processed as a single batch.245
The experts recognise that their domain of expertise is not the development of biotechnology techniques in the 1970s-80s and plant construction. They will share their own experience.
The manufacture of proteins in general and of plasma proteins in particular is characterised by high capital costs, complex manufacturing processes, and a considerable regulatory burden. Preparing PDMPs takes seven to twelve months between donation and final product release. The materials, equipment, plant, and resources required depend strongly on the number of different PDMPS produced, the continuity of supply of plasma and the scale of production.
Intermediate Cohn fraction production, protein purification including virus inactivation/removal steps, and filling may be carried out in different plants. The manufacturing process contains different production steps and filling vials (for small volumes of product such as Factor VIII concentrate) or bottles (for albumin) is one of the last ones. Strict compliance with Good Manufacturing Practices, quality assurance guidelines and regulations is required.
Fractionation technology is still changing because of new technological developments, new products, and safety requirements at different fractionation centres but most fractionators have adopted a cold ethanol method followed by chromatography and filtration steps as a matrix for the preparation of bulk plasma derivatives.
Below is a (non-exhaustive) list of materials/equipment used (excluding standard small materials), whose pharmaceutical quality must be adequate.246,247,248,249,250,251,252,253
These different types of filtration are characterised by their capacity to remove undesirable particles present in the products.
Depth filters are made with a multilayer porous medium capable of capturing contaminating particles throughout its bulk by a specific binding via weak electrostatic forces. Depth filtration and ultrafiltration are not absolute filtrations.
Since the 1980s, depth filtration has been carried out with filter aids such as AEROSIL (silica), used both as a filter aid and a centrifuge aid, and Celite (diatomaceous earth), used as a filter aid and to capture contaminants. Both of these play a role in eliminating impurities. Today, resin-bound asbestos is not used because of its toxicity.
Ultrafiltration (e.g. through 30- to 100-kDa membranes) is used to recycle the filtrate, concentrate proteins, and remove low-weight components (ethanol, salts). This process is much more effective than dialysis.
As plasma products cannot be sterilised in the final container, a sterilisation membrane must be used for final aseptic dispensing. The cut-off of this membrane has decreased from 0.45 µm in the 1970s to 0.22 µm and then to 0.2 µm (recently 0.1µm) so that the sterilisation of the product is improved and takes place under very clean manufacturing conditions.
Manufacturing facilities should adapt to the size of the production, with strict segregation of the area where donations (bags) are handled (a more “non-pharmaceutical clean” area) from the areas devoted to further process steps.
Treatment of waste, particularly ethanol and proteins, should be considered in the context of environmental protection.
Since the discovery of cryoprecipitate in the mid-1960s, different methods were used to remove partly the bulk of accompanying proteins, mainly fibrinogen, and to produce FVIII concentrates of intermediate-purity and high-purity.
Concentrate purity was increased both as a safety measure to reduce viral burden in the final product but also to reduce the putative risk of immune suppression. Factor VIII concentrates were classified according to purity and to the type of manufacturing procedure. The term purity refers to specific activity – the product’s content of Factor VIII coagulation factor (FVIII:C) in IU/mg (International Units per milligram) protein. In many concentrates specific activity is not a good measure of purity as albumin, for example, is added as a protein carrier and the von Willebrand Factor may be considered an impurity.
In general, intermediate-purity products have less Factor VIII activity per millilitre, which means that a larger volume has to be infused to obtain a specific number of units required for therapy; product purity thus affects the convenience of the treatment.
The recovery of Factor VIII coagulant activity (FVIII:C) is low in most fractionation procedures due to the lability of FVIII:C and the need to isolate other components from the plasma. Cryoprecipitation usually yields recovery of 300-500 units out of every 1,000 units theoretically present in 1 litre of starting plasma. Further purification to a high purity concentrate causes additional substantial loss. Therefore in routine practice, final recovery of 100-200 IU/L is normal which means that about 80% of the potential supply of FVIII:C is spoiled. Despite this, cryoprecipitation is still generally applied as the first purification step in Factor VIII production, because of the simplicity of the process and given that other plasma proteins can be isolated from the cryosupernatant. Although considerable literature has been published on the influence of several parameters on the recovery and purity of FVIII:C in cryoprecipitate, the mechanism by which the Factor VIII complex precipitates during freezing and thawing of plasma is not yet fully understood.254
Various measures have been tried in an attempt to improve the recovery of cryoprecipitate Factor VIII from plasma. FVIII could be lost at any step if delays were allowed to occur. It was recognised that freezing and thawing the plasma could improve the yield, for example by freezing the plasma in an extra-large bag.255
The yield was further optimised by the Australian thaw-siphon method, in which thawed plasma was continuously siphoned off during the thawing process.
In Canada, the use of heparin as an anticoagulant was demonstrated to improve recovery but the method became unpopular.256,257
At the stage of plasma collection, attempts were made to increase the Factor VIII level of the donor blood. Repeat plasmapheresis donors with high levels of Factor VIII were selected or donor plasma levels were increased by prior administration of DDAVP.258
The classification of purity shown in Table 4 is often used.
|
Preparation |
Manufacturing procedure |
Specific activity |
|---|---|---|
|
-Intermediate-purity -High-purity |
Cryoprecipitate + further purification Protein precipitation + chromatographic separation |
1-50 50-200 |
|
-Monoclonal antibody purified |
Affinity purified with monoclonal antibodies against FVIII or vWf |
> 1,000 |
|
-Recombinant |
DNA technology. Purification includes an affinity step with monoclonal antibodies against FVIII |
> 1,000 |
Low purity products refer to Fraction I and cryoprecipitate. Intermediate-purity concentrates are prepared from cryoprecipitate and further purified with a variety of methods, including Al(OH)3, glycine or polyethylene glycol precipitation. High purity products are prepared by conventional chromatography, and affinity chromatography with immobilised monoclonal murine antibodies directed against Factor VIII (FVIII) or von Willebrand Factor (VWF).259
FVIII concentrates are heterogeneous preparations where Factor VIII molecules represent about 1% of the total. The dose of Factor VIII cannot be expressed simply in milligrams. The lot potency of Factor VIII activity (FVIII:C) is designed. Potency is a measure of drug activity expressed in terms of the amount required to produce an effect of given intensity. Nevertheless, clotting factors are produced in licensed facilities and each lot of product is assayed for clotting factor activity during the production cycle and designed with a specific potency.260 This lot potency designation is needed for dosage calculations and is used by the prescribing physician for prophylaxis (for the prevention and management of haemorrhagic episodes) as well as during and after surgery. Factor VIII is assayed by one-stage and two-stage clotting methods and by chromogenic methods.
This is a technical question about FVIII activity determination. The difficulty is that Factor VIII is a co-factor and an indirect methodology should be applied.
There are two methods, the one-stage and the two-stages, that can be used in the diagnosis of haemophilia A or B, to classify disease severity, for potency labelling of FVIII concentrates by manufactures, to monitor post-infusion activity levels of FVIII during treatment and to test anti-FVIII antibodies (inhibitors).
In a simplified way, the one-stage activity (aPTT) assay measures the functionality of the coagulation pathway: determination of the time required for clot formation that is dependent on coagulation pathways. Normal aPTT values are usually within the range of 22-40 seconds.
The two-stage chromogenic factor activity assay measures the activity of Factor VIII in plasma. During the first stage, the test plasma or test Factor VIII concentrate is mixed with appropriate reagents and substrate (Factor X, phospholipids, CaCl2, prothrombin or thrombin), resulting in the rapid activation of Factor X to Factor Xa. The amount of Factor Xa generated is proportional to the level of functional factor under investigation.
During the second stage, a chromogenic substrate specific to Factor Xa and Factor Xa concentration is measured by monitoring the cleaved colored substrate. The colour intensity is proportional to the level of Factor Xa generated, which in turn is proportional to functional Factor VIII in the test.261
The methods should be calibrated against the WHO Factor VIII concentrate standard. For most plasma-derived Factor VIII concentrates, the results are similar using these assays except for products purified by immunoaffinity.262 Thus, the administration of an equal Factor VIII potency in units means the administration of different amounts of Factor VIII protein.263 Potency of FVIII/C is and should be estimated by in vivo pharmacokinetic studies and according to clinical efficacy.264
Similar conclusions could be drawn for FIX concentrates.265
The following section sets out how fractionation processes have developed over the past 40 years. It focuses in particular on the development of robust safety tests and new products (PDMPs) for clinical therapy, including detail on the medical uses of a number of different products in replacement and immune modulating therapy.
Over the last 40 years, viral reduction technologies have evolved from ‘no’ treatment to ‘multiple’ virus inactivation treatments.
In addition to the introduction of Good Manufacturing Practices (GMP), a robust net of safety measures was introduced for the different processes between donor and patient, under the supervision of Regulatory Authorities. The net of safety measures starts with epidemiological surveillance of the donor population in order to detect the prevalence of known transfusion transmitted infections (TTI) and the potential presence of emerging agents (Table 3). Risk groups are identified and if necessary certain groups are excluded from donation, or criteria for the restriction of donation are issued. In order to identify general risk factors, donors are screened for the occurrence of (potential) TTIs.
After donation, each unit is serologically tested for the presence of known pathogens such as hepatitis B virus, hepatitis C virus and HIV. Subsequently, mini-pool Nucleic Acid Technology (NAT) testing on known pathogens HIV, HBV, HAV, HCV and Parvovirus B19 takes place to guarantee a minimal infectious load in the manufacturing plasma pool. The risk of using contaminated units has been significantly reduced over time. Quantitatively, these improvements in final product safety margins have been in the range of about 100 fold for HBV, and 10,000 fold for HIV.266
The donations are then stored in quarantine for a period, so that if a donor has seroconverted, the putative contaminated donation can be withdrawn.
During the manufacturing process, the plasma pool is tested by NAT technology for known viruses such as HBV, HAV, HCV, and HIV. West Nile virus, dengue, chikungunya virus and Zika virus are also inactivated or removed by at least two viral inactivating/removing techniques. Dedicated virus inactivation and/or removal treatments are of crucial importance during the fractionation process in order to reduce the risk of emerging pathogens. The criteria for the viral reduction methods are a broad spectrum of inactivation and/or removal efficiency, optimal protein recovery and absence of protein denaturation. In the 1940s, pasteurisation became available to protect albumin-treated patients from hepatitis B transmission. Since the 1970s to 1980s, plasma fractionation has been at the forefront of development of viral reduction treatments. While developments prior to that were in response to existing virus threats, today, strategies focus on safeguarding against future emerging virus threats. Several viral reduction methods (inactivation and removal) used in plasma fractionation were developed during these years. In the 1980s, low pH in immunoglobulin products offered protection against enveloped viruses. In 1983, dry heat treatments were introduced in coagulation factor products to protect against HIV. In 1987, solvent detergent treatment became available against HIV, HBV and HCV in almost all products. In 1991, nanofiltration to discard large enveloped viruses was introduced, and in 2,000 caprylate acid technology demonstrated good viral inactivation in immunoglobulin products.
Fractionated plasma products have never been safer than today. When “orthogonal” complementary virus reduction treatments (which are independent of each other) are implemented, current plasma products will not transmit emerging viruses. Safety against emerging viruses should be built into the manufacturing process and new emerging viruses are inactivated or removed by existing technologies, through destruction of the viral envelope (S/D treatment) or the incompatibility of the size of the virus with the pore size of the filtration (nanofiltration). Careful pharmacovigilance monitors the efficacy and safety of the PDMPs (plasma derived medicinal products) in patients after their administration (also, Q10 and Q11).
The therapeutic functions of PDMPs are diverse. Many patients with a variety of diseases and disorders benefit from treatment with PDMPs. Due to the fact that plasma proteins have an important function in the wellbeing of the human body, therapy with a PDMP can be lifesaving and important in reducing morbidity and mortality. PDMPs have a therapeutic effect both in the prevention and treatment of disease, particularly for patients with coagulation and bleeding disorders, immunological disorders, infectious diseases, metabolic diseases and trauma. The World Health Organization has underlined the importance of PDMPs for global clinical care by putting a number of PDMPs on the WHO Model List of Essential Medicines. Anti-D immunoglobulin, anti-rabies immunoglobulin, anti-tetanus immunoglobulin, normal immunoglobulin for intramuscular, intravenous and subcutaneous use, coagulation Factor VIII and coagulation Factor IX are designated as essential medicinal products and governments “should make sure that all people can get access to these medicines they need, when and where they need them, which is vital to countries’ progress towards universal health coverage”.267
At the risk of oversimplification, therapeutic treatments with PDMPs can be divided into four categories: replacement or substitution therapies, immune modulating therapies, therapies directed at plasma protein antagonist functions, and anti-inflammatory therapies. Finally, some plasma proteins are under study for drug delivery, for example for acting as a carrier of a chemotherapeutic medicine in cancer treatments.
Replacement or substitution therapy refers to a PDMP being administered in patients with primary (congenital) or secondary (acquired) plasma protein deficiencies in whom the concentration of the protein concerned is absent or has become too low so that an adjustment of the plasma level is required. Depending on the plasma protein, an absent or too low concentration might be life threatening or seriously disabling, resulting in increased morbidity and mortality. Replacement therapy with the required plasma protein product is needed in such situations.
In the field of haemostasis, some patients are born with a quantitative or qualitative deficiency of a coagulation factor and replacement therapy is a therapeutic solution for overcoming coagulation disorder. These disorders are mostly rare chronic diseases, hereditary in origin, with an incidence of less than 5:10,000 and requiring lifelong treatment. Other patients acquire a factor deficiency due to an auto-immune response or as a complication of cancer. The best-known diseases of this type are haemophilia A (congenital, in primarily male patients, and acquired Factor VIII deficiency) and haemophilia B (congenital Factor IX deficiency). Depending on the level of endogenous factor, patients have a risk of life-threatening cerebral bleeds and of disabling bleeds, primarily in joints and muscles, if not prophylactically or on demand treated with cryoprecipitate, Factor VIII or Factor IX concentrate.268
Another factor deficiency is von Willebrand disease, which is caused by a deficiency of von Willebrand Factor and affects both men and women. Plasma derived von Willebrand Factor concentrate is used as replacement therapy for patients with von Willebrand disease type III. Purified plasma-derived concentrates of von Willebrand/Factor VIII are also used for treatment of bleeds and for surgical prophylaxis, when DDAVP (deamino D-arginine vasopressin) (the first treatment choice) is ineffective or contraindicated.269
Prothrombin Complex Concentrate (PCC) or PPSB containing Factor II, Factor VII, Factor IX with Factor X (4F) or without Factor VII (3F) is indicated for replacement therapy of isolated or combined congenital deficiency of (one of) these coagulation factors. These factors may, however, also be offered for treatment in a concentrated form as a single factor concentrate. PCC is also used in liver diseases with a too low concentration of clotting Factor II, VII, IX and X and when a correction is needed due to a (threatening) bleed or surgery. Factor VII and Factor XI are used to treat bleeds prophylactically, and are used on demand in patients with congenital or acquired deficiency of the relevant clotting factor. Factor XIII concentrate is indicated for congenital deficiency of Factor XIII, which, if untreated, results in haemorrhagic diathesis, haemorrhages and disturbances in wound healing. Fibrinogen (Factor I concentrate) is indicated as replacement therapy in haemorrhagic diathesis in the case of congenital afibrinogenaemia, dysfibrinogenemia and hyperfibrinogenaemia. Fibrinogen is also indicated in acquired hyperfibrinogenaemia due to diffuse intravascular coagulation (DIC) and hyperfibrinolysis in patients who do not respond to Fresh Frozen Plasma (FFP) and who do not react to other measures intended for correction of the fibrinogen consumption and the underlying cause. Fibrin sealant, which consists of the two components fibrinogen and thrombin, is indicated for creating a fibrin clot.
Some proteins have a role in the prevention of thrombosis and protein C is indicated as replacement therapy for severe congenital protein C deficiency. This product is also administered for the prevention and treatment of venous thrombosis, and purpura fulminans.
Plasma proteins have many functions in the homeostasis of the body. Due to the physiological properties of albumin such as maintaining the oncotic pressure, its transport and binding function and its scavenger effect in removing radicals, albumin is indicated for restoration and maintenance of circulating blood volume where there is a volume deficiency. Albumin is available in two concentrations: albumin with 40g/L or 50g/L and albumin with 200g/L or 250g/L, and each concentration has its own specific indications. Albumin 4% and 5% is indicated for the treatment of existing or threatening shock for example in the case of serious bleeds or burns, for extracorporeal circulation, sepsis or serious infections accompanied with serious protein loss (peritonitis, mediastinitis), transient arterial hypotension during haemodialysis and plasmapheresis or plasma exchange. Albumin 20% and 25% is indicated for ascites in combination with paracentesis, the treatment of hyperbilirubinemia in neonates as adjuvant, nephrotic syndrome in case diuretics do not have sufficient effect, acute liver insufficiency, serious postoperative hypo-albuminaemia and serious hypo-albuminaemia in neonates. Between 1998 and 2003, a number of studies led to scientific discussions on the safety of albumin compared to alternative treatments such as artificial colloids and crystalloids and whether albumin is associated with increased rate of death. The latest outcome was that although albumin has been determined to be safe for use as a resuscitation fluid in most critically ill patients and may have a role in treating early sepsis, its use is associated with increased mortality among patients with traumatic brain injury.270,271,272,273
Of the alternative treatments, the use of hydroxyethyl starch (HES) solutions is associated with increased rates of renal-replacement therapy and adverse events among patients in the intensive care unit. There is no evidence to recommend the use of other semisynthetic colloid solutions.274
Alpha1-proteinase inhibitor is used as replacement therapy for α1-antitrypsin deficiency, a genetic disorder that causes defective production of α1-antitrypsin leading to decreased α1-antitrypsin activity in the blood and lungs and deposition of excessive abnormal α1-antitrypsin protein in liver cells. Severe α1-antitrypsin deficiency causes panacinar emphysema or Chronic Obstructive Pulmonary Disease (COPD) in adult life in many people with this condition, especially if they are exposed to cigarette smoke. Unfortunately, the expansion of the α1-antitrypsin market is hampered by the absence of regulatory approval in many countries, lack of clinical trials demonstrating evidence-based therapeutic efficacy, inadequate diagnosis, limited awareness in the medical community and in the general public, and the high cost of therapy.275
Antithrombin III concentrate is indicated for the prophylaxis and treatment of thromboembolic complications in congenital or acquired anti-thrombin III deficiency, a rare and under-recognised medical condition associated with inadequate endogenous anticoagulation.276
C1-esterase inhibitor concentrate is indicated as substitution therapy for the prevention and acute treatment of attacks of oedema in primarily the throat, extremities and abdomen caused by hereditary angioedema, a congenital or acquired C1-esterase inhibitor deficiency. In an attack of oedema in the throat area, the oedema can block the trachea and, if untreated with lifesaving C1-esterase inhibitor concentrate, the patient may suffocate.277
In 1952, Bruton published his classic description of congenital agammaglobulinaemia and following this publication the intramuscular administration of immunoglobulin concentrations became regular practice.278
Patient groups with primary and secondary immune deficiency are dependent on immunoglobulin G (IgG) concentrate which protects them against life-threatening infections, and polyvalent intravenous immunoglobulin (IVIg), intramuscular immunoglobulin (IMIG) and subcutaneous immunoglobulin concentrate (SubIg) are indicated to replace the missing protein. IgG products are indicated for primary immune deficiency syndromes and/or disorders in specific antibody formation due to congenital a-gammaglobulinaemia and hypoglobulinaemia, common variable immune deficiency (CVID), serious combined immune deficiency (SCID), Wiskott-Aldrich syndrome, DiGeorge syndrome, ataxia-telangiectasia, and IgG-subclass deficiency.
In secondary immune deficiency syndromes, IgG products are indicated for chronic lymphocytic leukaemia, children with AIDS, allogeneic bone-marrow and other forms of transplantation, and preterm babies with a body weight at birth of less than 1,500 grams. Hyper-immune globulins or specific globulins, mostly administered intramuscularly, are immunoglobulin concentrates manufactured from donor plasma with a high titre of specific antibodies. Hyper-immune plasma is collected from donors who have been actively immunised with specific antigens such as Anti-D, rabies, tetanus, hepatitis B, anthrax and smallpox. Hyper-immune plasma can also be obtained from donors with naturally occurring antibodies such as cytomegalovirus (CMV), varicella (VZV), hepatitis A or respiratory syncytial virus (RSV). Hyper-immune globulin preparations are of great importance for the prevention and mitigation of life-threatening infections with the pathogens concerned if the persons are not vaccinated, if vaccination is not possible, or in whom the vaccination has not resulted in a sufficient protective antibody plasma level. Intramuscular immunoglobulin (IMIg) is also indicated for passive immunisation against hepatitis A.
When first in clinical use in the 1950s, immunoglobulins were administered only by the intramuscular route because serious adverse events had occurred after an intravenous injection, caused by activation of the complement system due to aggregation of IgG. It took almost 20 years after Barandun’s development of an intravenous preparation of IgG for the administration of IVIg in 1962 for it to become common practice.279
The observation of Dr Paul Imbach that the therapeutic value of IVIg could be associated with effects other than the passive transfer of antibodies alone, completely changed the IVIg treatment options.280 In the treatment of two immune deficient children suffering simultaneously from serious immune thrombocytopenia, he noticed that the platelet count increased after administration of a high dose of IVIg. This observation was confirmed by others and following the results with ITP, high doses of IVIg have been administered in patients suffering from many other immune-haematological disorders, autoimmune diseases, neurological syndromes and other diseases of the immune system. Some of these reports are only case reports; others are the result of randomised clinical trials. The results do not always match with other studies, which can be explained by the fact that the pathophysiology of the disease and the mechanisms which could explain the therapeutic effect of IVIg, have not yet been fully understood.
Currently, high dose IVIg is indicated for Idiopathic Thrombocytopenia (ITP), Guillain Barré syndrome, Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) and Kawasaki’s disease. Some IVIg products can also be used to treat Multifocal Motor Neuropathy (MMN), dermatomyositis, autoimmune uveitis and myasthenia gravis. Since the introduction of high dose IVIg, the need for polyvalent intravenous immunoglobulin has grown significantly for the treatment of an increasing number of haematological, neurological and dermatological auto-immune diseases such as auto-immune limbic encephalitis, stiff person syndrome, autoimmune haemolytic anaemia, post bone marrow transplantation, parvovirus B19-associated aplasia, toxic epidermal necrolysis, Stevens-Johnson syndrome, and immune bullous disease. Both in Cochrane meta-analyses for isolated indications and national and international consensus meetings on multiple indications, efforts are being made to classify the indications for immune modulating therapy of IVIg in order to establish evidence-based usage and to minimise off-label use.281,282,283,284
Prothrombin complex concentrate (PCC) containing Factor II, Factor VII, Factor IX and with Factor X (4F) or without VII (3F) is indicated for reversal of Vitamin K antagonist treatment. The vitamin K-antagonist function of PCC is stronger than fresh frozen plasma and also lifesaving.285
An activated PCC Anti-Inhibitor Coagulant Complex or Factor Eight Bypassing Agent-FEIBA is a Factor VIII inhibitor bypassing agent to control spontaneous bleeding episodes and is used in surgery in haemophilia A patients with inhibitors.286 This function is quite important given that inhibitor formation is currently the major adverse effect of haemophilia treatment.287
The anti-inflammatory actions of IVIg are mainly based on the hypothesised working mechanisms of immune modulation in which IVIg in a high dose has been shown to be efficacious. Intravenous immunoglobulin is manufactured from more than 1,000 donations and contains more than 2 million antibodies. For that reason, it is hypothesised that IVIg may derive its immune modulating effect from an anti-inflammatory function. A number of the approved indications are correlated with a pre-existing infection. For example, in 25% of the cases of Guillain-Barré syndrome a correlation with infection with Campylobacter Jejuni has been shown.288 A survival benefit was observed for patients with sepsis who received polyclonal intravenous immunoglobulin therapy compared with those who received a placebo or no intervention.289 More importantly, in research into the immune modulating working mechanisms of high dose IVIG, the role of its anti-inflammatory properties in innate immunity is becoming more prominent.290,291
Factor VIII is an essential, non-enzymatic, blood-clotting protein also known as anti-haemophilic protein. In the bloodstream, it circulates in a stable, non-covalent complex with von Willebrand Factor until an injury that damages blood vessels occurs. Factor VIII acts as a cofactor of Factor IX in the activation of the coagulation and the formation of the clot.
In the bloodstream, when activated by thrombin, Factor VIII cleaves to generate separate A1 and A2, A3-C1-C2 chains and the release of B-domain and the von Willebrand Factor, generating a family of heterodimers.292
Factor VIII is an unusually large protein containing 2,332 amino acids synthesised as a glycosylated single chain with a molecular weight of about 330 kDa. Factor VIII contains three different domains, an A-domain that is repeated three times (A1, A2, A3), a central B-domain and a repeated C-domain (see scheme Figure 7). There are small peptide regions, one of which joins the A1- and A2-domains and two other joining the B-domain, with the A2-domain and on the other side with the amino-terminal of the A3-domain.293
Factor IX circulates in blood as a small, glycosylated single-chain protein with a molecular weight of approximately 56 kDa. When a vessel injury occurs, FIX is activated and interacts with other coagulation factors and platelets in a series of reactions that lead to blood clotting.294
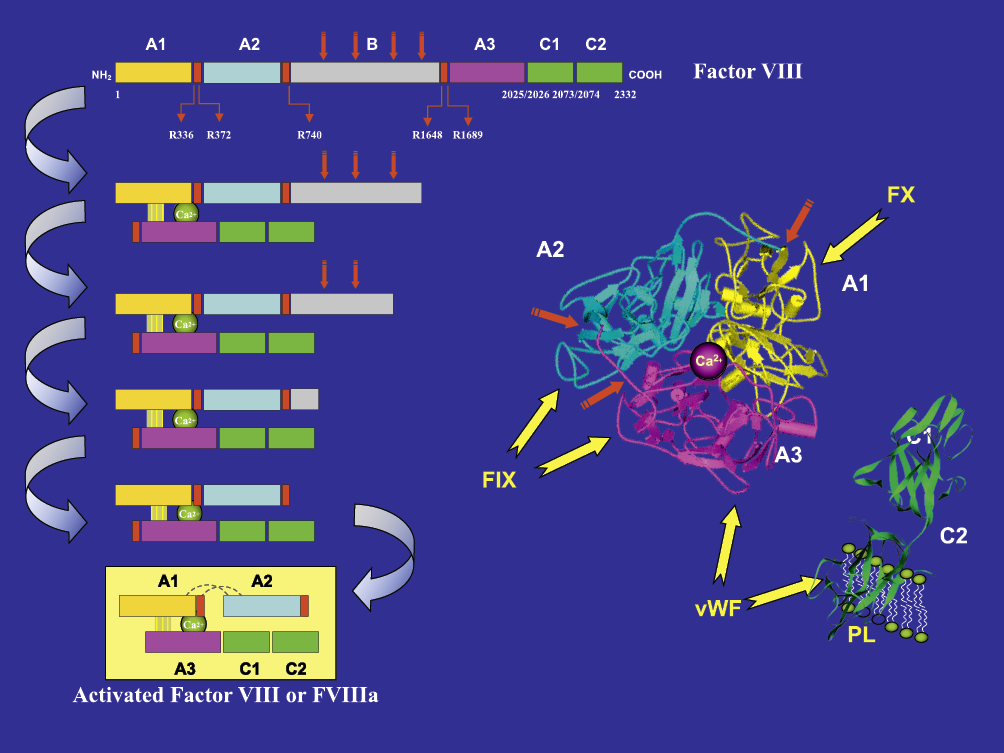
Left: Factor VIII structure and proteolytic processing of Factor VIII into activated Factor VIII.
Right: Binding sites of Factor VIII with Factor IX (FIX), Factor X (FX), von Willebrand Factor (VWF) and Phospholipids (PL) as indicated by yellow arrows. Domain C1 (in green) is behind domain C2.295
Factor VIII is usually administered to patients intravenously, a normally tolerogenic route to safely administer foreign proteins, yet is highly immunogenic compared to many other therapeutic proteins (see also section 9B.4.ii).296
Prophylactic or on-demand treatment of bleeds with recombinant or plasma-derived Factor VIII can lead, unfortunately, to an antibody response to this human but “foreign” protein that effectively neutralises or inhibits its function in the coagulation pathway. These antibodies are called “inhibitors” and recognise accessible functional Factor VIII peptide sequences (epitopes). Today, the role that the presence of inhibitor and non-inhibitory antibodies (specific immunoglobulin) in healthy donors, plasma pools and people with haemophilia, may play in tolerating Factor VIII is the subject of research.297 It is hard to find the cause of the formation of anti-Factor VIII antibodies as it is a multifactorial complex phenomenon in which genetic and environmental factors interact dynamically. Today, the development of inhibitory antibodies is the most serious and challenging complication of haemophilia therapy. The consequence of development of inhibitors in people with haemophilia is difficult-to-control haemostasis, especially during acute bleeding episodes and elective surgery.298,299
Inhibitor development in haemophilia B is rare due to the fact that, in contrast to haemophilia A, most patients with haemophilia B have a dysfunctional form of Factor IX.
In many countries a variety of FVIII concentrates are now available, both plasma-derived and recombinant products. They differ in the manufacturing process, the type of virus inactivation used and in purity. Beside the aspects of efficacy, purity, availability, cost and convenience of handling, the most important criterion in the choice of a concentrate is viral safety as a consequence of the devastating sequelae of the AIDS and hepatitis epidemics. In consequence, Factor VIII production processes have to assure effective and safe clotting factor concentrates.
Antibodies or inhibitors to Factor VIII may arise as allo-antibodies in haemophilia patients who have been transfused with exogenous recognising Factor VIII (see also 9E, 1, ii).
There has been an ongoing debate regarding the immunogenicity of Factor VIII products with a concern that recombinant products have an increased risk of inhibitor formation over that of plasma-derived products.300
Current regulations require use of at least a combination of two effective orthogonal virus reduction steps to target infectious agents, based on different principles of action.301,302
PDMPs manufacturers introduced virus-inactivation steps in the existing Factor VIII production processes. One of the leading viral inactivation/removal technologies is pasteurisation, involving heating to 60°C for 10 hours. This has been used effectively for human plasma albumin for more than 50 years and was introduced as a new production step in the Factor VIII production process. However, clotting proteins are heat-labile in liquid-state and fractionators tried to minimise protein denaturation by adding stabilisers or protective agents as sugars, polyols and amino-acids that must be removed in subsequent process steps (see also Question 10).
Two plasma-derived Factor VIII concentrates produced by two different manufacturers (Factor VIII CPS-P, CLB, Amsterdam, The Netherlands and Bisinact, Octapharma, Vienna, Austria), both treated by a new pasteurisation virus inactivation method, have been associated with a sudden increased development of Factor VIII inhibitor after their infusion in patients.303 The rise of inhibitors in these patients, who had been exposed to Factor VIII concentrates for more than 50 days, is a rare event. This event alerted the medical community worldwide to the possibility of inhibitor formation following treatment with new virally inactivated products. In both cases, to increase virus safety a new pasteurisation step was introduced in well-known FVIII concentrate production processes recognised as not associated with inhibitors.
The first therapeutical concentrate, Factor VIII CPS-P, was a FVIII concentrate that was purified by controlled-pore silica adsorption and was not associated with inhibitors. To inactivate viruses, a new pasteurisation at 60°C for 10h step was introduced in the Factor VIII production.304 Sixteen previously treated haemophilia A patients in Belgium and in the Netherlands developed inhibitors after exposure to Factor VIII CPS-P. Barrowcliffe showed that the new pasteurisation process might have induced activation of Factor VIII (Figure 7).305 Further studies showed that the patients’ inhibitors mainly recognised the FVIII C2-domain (see also section 9E.1.i-ii).306
A second well-documented occurrence of such inhibitors in patients was found with a Factor VIII concentrate (Bisinact) treated with solvent/detergent (S/D) (active on enveloped viruses) followed by a new additional pasteurisation step at 63°C for 10h to inactivate the non-enveloped viruses.307 Eight out of 140 multi-transfused Belgian haemophilia A patients developed inhibitors after changing treatment to double-virus inactivated plasma-derived factor concentrate. The inhibitors gradually declined when exposure to the S/D and pasteurised product was stopped, despite further treatment with other Factor VIII treatments. The inhibitors were found to be specific to the Factor VIII light chain.
Inhibitors also appeared in 12 out of 109 German patients treated with a similar product (Factor VIII concentrate treated by S/D followed by a pasteurisation at 63°C for 10h). The epitope specificity of 8 German inhibitor plasmas was also found to be restricted to the Factor VIII light chain, in particular to the C2 domain.308
Further studies of this concentrate suggest that the combination of S/D and pasteurisation at 63°C may enhance exposure of the phospholipid binding site in the C2 domain of Factor VIII, and since inhibitors in this product are predominantly against C2, these findings could be relevant to the enhanced immunogenicity of the concentrate.309
In 1987, pre-licensure clinical trials with the first recombinant Factor VIII products began in patients with haemophilia A. In the PUP (previously untreated patients) trials inhibitor antibodies developed early – after a median of 9-11 exposure days (EDs) – in 20-25% of the study subjects. Approximately half of the inhibitors in both PUP studies were ‘high-responders’ (> 5 Bethesda units) whereas the remainder were ‘low responding’ and most of these inhibitors were transient. Nevertheless, some clinicians became concerned that recombinant FVIII was causing a higher incidence of inhibitors. However, studies in infants and children with severe haemophilia A published in 1992-1993 documented a higher incidence of inhibitor development with plasma-derived FVIII (25-50%). Prospective studies with monitoring inhibitors at frequent intervals, showed that 25-35% of previously untreated patients would develop inhibitors after a median of 9-11 EDs. Other analyses were documenting that patient-related factors such as severity of haemophilia, ethnicity, and FVIII gene mutation causing the person’s haemophilia, were more important determinants of inhibitor development. However, it became apparent that very few persons with severe haemophilia who had received > 50 EDs with plasma-derived FVIII developed de novo inhibitors while on recombinant FVIII. These findings led to the 1999 recommendation by the Scientific Subcommittee on FVIII and FIX of the ISTH’s Scientific and Standardisation Committee that the evaluation of the immunogenicity of any new FVIII product be carried out in previously (and heavily) treated haemophilia patients.310,311,312,313,314
The most important side effects of treatment with clotting factors include the induction of alloantibodies, modulation of the immune system, thrombotic events and viral transmission. In addition, there can be minor adverse reactions following treatment with factor concentrates, such as rashes (urticaria), flushing and erythema (rash caused by capillary congestion) at the infusion site.
As mentioned before, the development of antibodies to Factor VIII in some patients, so-called inhibitors, is a major complication in haemophilia treatment. Following treatment with plasma-derived clotting factor concentrates approximately 10% of haemophilia A patients develop alloantibodies to Factor VIII whereas the percentage of inhibitors in haemophilia B patients is about 1-4%. Inhibitors to Factor VIII and Factor IX usually develop within a limited number of exposure days.315
Inhibitors have been estimated to occur in 20% of all patients with severe haemophilia A, although one study found an even larger risk of 52% in severe haemophilia A. Because bleeding cannot be effectively treated or prevented by Factor VIII administration, inhibitor formation significantly complicates therapy. Inhibitor patients cannot fully enjoy the benefits of substitution therapy, and their risk of mortality is higher than that of patients without inhibitors. Inhibitors usually develop in the early phase of treatment with clotting-factor preparations, in many cases after only a few infusions. Once more than 100 to 250 infusions have been administered, inhibitor development appears to be rare and inhibitors are often detected at a young age, but occasionally, transient inhibitors are detected in older multi-transfused patients, usually without clinical consequences. Developments since the 1960s have led to the availability of more and more concentrated Factor VIII products: Factor VIII concentrates purified by monoclonal antibodies (MoAbs) and concentrates produced by recombinant DNA techniques. Concerns have been raised that these ultra-pure concentrates might entail a higher risk of inhibitor development compared with conventional concentrates.
There is a genetic predisposition to the development of inhibitors, such as a large deletion of the Factor VIII gene. Inherited factors, family history, ethnicity, environmental factors, the type of concentrate, and treatment early in life with high doses contribute to the development of inhibitors. There is some evidence that plasma-derived Factor VIII concentrates containing von Willebrand Factor are less immunogenic.316
The UK Haemophilia Centre Doctors’ Organisation calculated a cumulative risk of inhibitor development of 16% by the age of 5 years and 36% by the age of 75 from data in 6,078 patients with haemophilia A, and a cumulative risk of inhibitor development of 6% by the age of 5 years and 68% by the age of 75 from data in 1,172 patients with haemophilia B.317 The risk of inhibitor development is at its highest within the first 50 days of exposure to the infused factor.318,319
As mentioned before, in 1993, an unexpected high prevalence of Factor VIII inhibitors was described after exposure to a pasteurised Factor VIII concentrate. All patients with more than 200 and up to 1,000 days of exposure to Factor VIII had been previously transfused with other Factor VIII concentrates and had tolerated this well. The mechanism for the unexpected high prevalence of these inhibitors is unknown.320,321,322
Treatment with recombinant Factor VIII induced antibodies to Factor VIII at a relatively high prevalence of 25%. The concentration of antibodies was relatively low and treatment with Factor VIII could be continued, although at a higher dose. Several alternative products are available for the treatment of patients with high titre inhibitors.323,324
One of these is porcine Factor VIII, which is used both for the treatment of patients with haemophilia who have developed alloantibodies to Factor VIII and in patients with acquired haemophilia due to auto-antibodies against Factor VIII. Adverse reactions associated with porcine Factor VIII are rare, with mild thrombocytopenia and anaphylactic reactions being reported. About 20% of patients with acquired haemophilia developed specific anti-porcine antibodies during porcine Factor VIII treatment.325
Treatment of inhibitors comprises immune-tolerant protocols with plasma-derived or recombinant Factor VIII, activated prothrombin complex concentrate or recombinant Factor VIIa.
Activated prothrombin complex concentrates such as FEIBA (Factor VIII inhibitor bypass activity) are used to treat haemophilia patients with Factor VIII inhibitors. As thrombogenic complications associated with FEIBA have been reported, it has been suggested that haemophilia B patients without inhibitors or with low titre inhibitors should be treated with Factor IX concentrate.326
Other products for the treatment of Factor VIII inhibitors are prothrombin complex concentrates and recombinant human activated Factor VII. Recombinant Factor VIIa has been used effectively and safely in haemophilia patients with inhibitors. However the product is associated with thrombotic events, possibly because of unbalanced activation of the coagulation cascade. In addition to mild adverse effects, disseminated intravascular coagulation has also been reported.327
In vivo and in vitro studies have shown that many people with haemophilia have immunological abnormalities particularly with regard to cell mediated immunity; these include low CD4 lymphocytes and abnormal CD4/CD8 ratio, and a decreased response of lymphocytes to stimulation with non-specific mitogens like phytohaemagglutinin and concanavalin A. Factor VIII and Factor IX concentrates exhibit a dose-dependent inhibitory effect on lymphocyte IL-2 secretion which may be explained by down regulation of CD25 expression or inhibition in early stage lymphocyte activation by one or more components in Factor VIII preparations.
Also, down-regulation of IgG Fc receptors on the monocyte surface and decreased monocyte functions have been noticed. It is suggested that immune complexes composed by blood group antigen and antibody may be implicated in the modulatory effect of concentrates on monocytes. The cause of death in people with haemophilia due to infections was 5% compared to 0.9% in non-haemophiliacs. When death due to acute hepatitis was excluded the mortality due to infection in people with haemophilia was still 3.3%.
Also, a report that 38% of a population of boys with haemophilia developed pulmonary tuberculosis is evidence for the fact that people with haemophilia are susceptible to infection. The incidence of 38% pulmonary tuberculosis in people with haemophilia was comparable with the incidence of pulmonary tuberculosis in immunocompromised patients.328
It has been suggested that intermediate purity products may adversely affect immune functions in recipients, independent of any alterations due to infection with HIV. This has led some physicians to advise that high purity products are preferable. However, in HIV positive people with haemophilia treated with high purity Factor VIII concentrate, the median decline of CD4 cells was similar to that in HIV positive people with haemophilia treated with intermediate purity Factor VIII concentrate, suggesting that high purity Factor VIII concentrate appears to have no apparent beneficial effect on the CD4 cell count.329
Anaphylactic reactions upon administration of coagulation factors are rare. When occurring, they are possibly associated with inhibitor development, especially in the case of Factor IX inhibitors.330,331
Deficiency of Factor IX is a less common bleeding disorder with an unpredictable bleeding tendency. Earlier reports of the use of Factor IX concentrate described anaphylactic shock and thrombogenic complications. In a study in which 30 patients were treated with a dry heated Factor IX concentrate, minor side effects occurred in only one patient who was known to have had allergic reactions after FFP treatment. In this study no thrombotic complications were observed.
Heat treatment of viruses is an effective sterilisation method. A virus particle can be considered truly inactivated if its reproductive capacity is totally and irreversibly destroyed through chemical alteration of its nucleic acid genetic material. Heat also denatures proteins by modifying their highly organised structure (folding-unfolding) and inhibiting both the virus penetration into the cell and its multiplication.
Nucleic acids, including RNA and single- and double-stranded DNA, are nucleotide polymers synthesised by polymerase enzymes during transcription or DNA replication. Along the backbone, individual nucleotides are capable of interacting with one another via hydrogen bonding, allowing formation of higher-order organised structures. Denaturation due to high temperature results in disruption of base pairs and alters the integrity of DNA and RNA. Double-stranded (ds)DNA separates into two component strands (it is said to melt). After cooling, renaturation of DNA requires good base pair matching.
The mechanism through which viruses are inactivated by heat can vary according to the temperature.332 At lower temperatures, inactivation can proceed through degradation of viral nucleic acid. At higher temperatures, destruction of the virus coat protein and of the cell-receptor-binding domain can occur. Heat treatment acts upon the virus’s capsid protein, strongly reducing viral infectivity towards host cells. Exposure to heat causes the protein to lose its native structure via unfolding and/or conformational changes that alter protein quaternary, tertiary, or secondary structure. Virus inactivation depends on not only the temperature, but also on the pH, the ionic strength, and the virus strain and structure. In the case of the polio virus, a single-stranded RNA virus, the mechanism of heat inactivation involves stepwise antigenic changes and eventual exposure of the viral nucleic acids without capsid disassembly. Viral egress is thought to occur through a small pore in the capsid. Loss of infectivity following heat treatment, the release of viral nucleic acid, and antigenic conversion occur simultaneously.
Heat likewise inactivates the non-enveloped human virus B19 (a single-stranded DNA virus) by converting infectious virions to DNA-depleted capsids.333 In contrast, other viruses such as the foot-and-mouth disease virus (a single-stranded RNA virus) show capsid disintegration accompanied by nucleic acid release.
Processes that inactivate viruses may also denature proteins of interest.334 Intermolecular bonds and solvent interactions determine the conformational stability of a protein. Alteration of the three-dimensional structure may be induced by temperature, by pressure during heating, and by the solvent used. Hydration of molecules is likely to play a major role in maintaining the native state. As high temperature destabilises hydrogen bonds and non-covalent bonds, it modifies protein tertiary structure. Unfolding can be, to some extent, reversible when the temperature is lowered. If a mistake in the folding mechanism occurs, changes may occur that will result in partial or complete inactivation (denaturation) of the proteins in general, and plasma proteins in particular.335
Protein thermal denaturation depends on the amino acid composition (e.g. the proline content). As PDPM preparations are not pure protein preparations (Factor VIII constitutes only 1% of high-purity FVIII concentrate), a reaction may occur with the accompanying proteins and this complicates prediction of the effects of a temperature increase. A protein may be stabilised by a ligand that rigidifies the molecular structure (e.g. caprylate stabilises albumin). Modifying a protein’s structure leads to its inactivation and reduced efficacy or even to its immunogenicity potential in patients.
There are two processes of viral reduction used in fractionation: inactivation (killing the virus) and efficient removal (clearance) of virus through purification of protein. The contribution of virus neutralisation by plasma specific antibodies to viral safety is recognised.
The potential risk of virus transmission by PDMPs in general, and by Factor VIII and Factor IX concentrates in particular, can be considerably reduced by including a heat treatment step in the manufacturing process. Heat treatment is recognised as an effective way to inactivate microorganisms, including viruses, in the vaccine and food industries. It is widely used by fractionators for a variety of plasma products. As previously mentioned, virus inactivation techniques based on thermal treatment aim to make viruses unable to infect cells and to multiply, thanks to denaturation of viral proteins through alteration of their three-dimensional structures followed by aggregation/fragmentation and denaturation of viral nucleic acids.
Ideally, a virus-inactivated product can be regarded as safe if the maximum quantity of product a patient receives contains less than the smallest infectious dose of virus.336 To provide evidence that the heat treatment is efficient, extensive validations must be performed to provide indirect evidence that the production process might inactivate or remove virus contamination with a panel of enveloped and non-enveloped viruses and the relevant HIV.337,338
Three basic strategies are used to thermally inactivate viruses and produce safe PDMPs: heating in solution (pasteurisation), vapour (steam)-treatment, and heating of dry products.339,340,341,342,343,344
Only well-established processes are reported here.
Pasteurisation is a virus inactivation method widely applied in PDMP production and effective against a wide variety of viruses. The effect of pasteurisation is process and product specific, and product stability is a limiting factor.345
Pasteur initially developed heat treatment in 1860. A heat treatment process developed during World War II to stabilise albumin in its final container in the presence of stabilisers is widely used by all fractionators. This process (60°C-10 h), usually performed in a tank filled with high-quality water, is known to inactivate the enveloped viruses, and non-enveloped viruses including the non-enveloped hepatitis E more recently identified.346 These conditions could not be implemented for other PDMPs without being adapted.
In 1981, Heimburger et al described a method to heat a Factor VIII preparation (Haemate P) in the presence of sucrose and glycine for 10h at 60°C. The FVIII preparation is then separated by precipitation. The efficacy of HBV inactivation was assessed in a study in chimpanzees. The yield is low, at about 8% of the initial plasma. The loss of yield due to the application of heat resulted in the need to obtain larger volumes of plasma.347,348
The pasteurised Factor VIII was licensed in 1981 under the name of Haemate HS in Germany (also called Haemate P), followed by the publication of two early clinical studies. One hundred and fifty-five eligible patients with haemophilia A or von Willebrand’s disease attending 11 haemophilia centres in the Federal Republic of Germany, were selected. Between February 1979 and December 1986, they received a total of 15,916,260 IU of pasteurised Factor VIII (60°C-10h) (Haemate HS, Behringwerke). By September 1988, the patients were tested for HIV-1 (all) and HIV-2 (66 patients). All were found negative. Neither hepatitis nor post-transfusion seroconversion for hepatitis in 26 haemophilia patients treated for one year with 32 batches of pasteurised-Factor VIII were found.349,350,351,352
The next major change to increase Factor VIII stability and yield, introduced in 1988, included the replacement of citrate with antithrombin and heparin during the purification process of Haemate.353
For PDMPs such as antithrombin and α1-antitrypsin, pasteurisation (60°C-10h) is performed in-process in the presence of high doses of stabilisers (sugars, amino acids or acetate, polyols) that must be eliminated in a further process step, mostly by ultrafiltration or chromatography.354,355,356
Pasteurisation inactivates enveloped viruses (e.g. HBV, HCV, and HIV), emergent viral pathogens and non-enveloped viruses, e.g. HAV, but is generally of limited value in the inactivation of parvovirus B19.357,358,359
Different immunoglobulin manufacturers use pasteurisation as a virus inactivation method at low pH and ionic strength. Heat treatment of immunoglobulin may induce polymer formation if the protein concentration is too high (about 5%), and changes in immunoglobulin secondary structure may occur.360 Therefore, specific excipients have been developed for IVIgs. For instance, one fractionator uses 33% (w/w) sugar (sorbitol) at pH 5.5 and heats to 60°C for 10 h.361
This is a two-stage process developed by the fractionator Immuno AG (now Takeda). This proprietary treatment is restricted to one manufacturer, and consists of generating steam (1,190 mbar) in a homogeneously wetted intermediate lyophilised product for 10 h at 60° followed by 1 h at 80°C. The water content has to be adjusted to 7-8% (w/w). The in-process treatment is carried out in a closed system under an inert gas.362,363 It inactivates enveloped viruses and some non-enveloped viruses such as HAV.364,365
While an abstract was published in 1980, a prospective and clinical safety study with steam-treated Factor VIII (24 patients) was published in June 1984.366
Terminal dry heating is a virucidal treatment, mostly performed on the final freeze-dried product. In light of the urgent need to interrupt the continuing transmission of AIDS by Factor VIII, the dry heating process was widely adopted.367 Heating is carried out at the last step of the process when the product is sealed in its final container, thus eliminating the chance of recontamination. This approach is, along with pasteurisation, one of the leading virus inactivation technologies used today.
Initially, dry heating at 60 to 68°C for up to 72 or 96 h was applied to coagulation factor concentrates to inactivate HIV. However, this failed to inactivate HBV and HCV in a chimpanzee model and clinical studies.368 Desiccation appears to stabilise not only Factor VIII but also the potentially contaminating viruses.369
Today two sets of conditions (“severe dry-heat”) are applied: 80°C for 72 h or 100°C for 30 min. Other conditions are described by manufacturers as 100°C for 120 min.370
Most Factor IX concentrates are subjected to terminal dry heat treatment. The prothrombin complex preparation, containing Factor IX, Proplex T and the partially activated prothrombin complex Autoplex (Hyland-Baxter), are both heated at 60°C for 144 h.371,372
The critical residual moisture content of the final product must be carefully controlled (between 0.8 and 2%).373,374 If the product is too dry, the virus inactivation efficacy may be reduced. If the moisture content is too high, Factor VIII may be affected.
The European Medicines Agency recommends that residual moisture should preferably be measured on each vial of final product using a non-destructive method.375 Terminal dry heating inactivates enveloped and some non-enveloped viruses such as HAV but parvovirus models (canine and bovine parvovirus) are more resistant.376,377 It is often included as a second inactivation method in the product process.
Table 5 shows the chronology of some heat-treated Factor VIII concentrates put on the market.
|
Method – Fractionator |
Date applied for FDA Licensing (*) |
Date License granted by FDA*, UK**, publication*** |
|---|---|---|
|
Dry-heat (60°C – 72/74h) (a) Baxter Healthcare (US) |
June 1982* |
March 1983* |
|
Pasteurisation (60°C – 10h) (a) Miles Inc (formerly Cutter Biological) (US) |
August 1983* |
January 1984* |
|
Wet heat (60°C – 20h) (a) Alpha Therapeutics (US) |
December 1982* |
February 1984* |
|
Dry-heat (60°C – 30h) (a) Armour Pharmaceutical (US) |
December 1982* |
January 1984* UK licence 1989** |
|
Terminal Dry-heat (80°C – 72h) (b) BPL (England) – SNBTS (Scotland) |
1985 – 1987 |
|
|
Pasteurisation (60°C – 10h) (b) Armour (US) |
UK licence 1989** |
|
|
Terminal Dry-heat (80°C – 72h) (b) CSL (Australia) |
Australia only** |
|
|
Terminal Dry-heat (100°C – 30min) (c) Shire (formerly Biotest) |
Germany 1996*** |
While heat treatment as a virus inactivation method was investigated in order to improve the safety of clotting factor concentrates, simultaneously the effect of this treatment on purity, potency and yield was the subject of research.
In order to evaluate the influence of heat treatment (68 °C for 24 or 72 hours) on the essential components of anti-haemophilic cryoprecipitate, i.e. Factor VIII coagulant activity (VIII:C), von Willebrand Factor (VIIIR:Ag – FVIII related antigen and VIIIR:RCF – FVIII Ristocetin cofactor) and fibrinogen, ordinary lyophilised cryoprecipitate was compared to heat-treated, amino acid-enriched specimens. The median reduction in Factors VIII:C, VIIIR:Ag, VIIIR:RCF and fibrinogen during lyophilisation of ordinary cryoprecipitate was 26%, 11%, 1% and 8.5% respectively. Heat-treatment of such cryoprecipitate resulted in 85 to 98.5% reduction in these parameters, while the reduction following lyophilisation and heat treatment (24 hours) of amino acid-containing preparations was not significantly different from non-heated, ordinary cryoprecipitate. Following heating of amino acid-enriched cryoprecipitate for 72 hours, only Factor VIIIR:RCF was significantly reduced (32.5%) compared to non-heated samples. For Benny et al., heating freeze-dried cryoprecipitate in the presence of Synthamin 17 (mixture of amino acids) at 60°C for 48 h allowed a FVIII recovery of 89%.381 Their method was routinely used and produced 1.6 million Factor VIII units per year.382 Finding favourable conditions to heat cryoprecipitate was mostly based on experimental research as most of the clotting factor activity was severely reduced after heating. The addition of Synthemin (mixture of amino acids) allowed the heating of cryoprecipitate and inactivates LAV/HTLV III (HIV) but not all pathogenic viruses.
Evaluation of the effects of dry heat treatment (96 h at 68° C) to eliminate LAV/HTLV-III virus on Factor VIII (FVIII) and von Willebrand Factor (VWF) present in an intermediate-purity concentrate showed that this thermal inactivation appeared to have little effect on FVIII. A loss (12.3 +/- 3.6%; n = 25) in FVIII coagulant activity (FVIII: C) and a good in vivo performance in haemophilia A patients was considered acceptable. A precise analysis of VWF indicated that whereas the VWF antigen and its ristocetin cofactor activity decreased during heating, there was an increase in potentially functional forms of VWF. Heat treatment induced an increase in high molecular weight forms of VWF and an enhancement in platelet adhesion to collagen. These changes probably explain the correcting effect on the bleeding time of the heated FVIII concentrate in patients with von Willebrand disease. Thus, heat-treated concentrate appears to be equivalent to the untreated product in haemophilia A, with the additional benefit of being efficient for the treatment of von Willebrand disease.383
When the pharmacokinetic characteristics of a high-purity pasteurised FVIII concentrate were assessed in comparison with an intermediate purity pasteurised concentrate produced by the same manufacturer, the outcome of the study was that the pharmacokinetic characteristics were very similar. The study was designed as a crossover single-dose pharmacokinetic investigation in 8 non-bleeding patients with severe haemophilia A. The concentration of Factor VIII was determined in triplicate in three different laboratories using each of the following assay methods: a one-stage clotting assay, a two-stage clotting assay and a two-stage chromogenic-substrate assay. The findings indicated that the purification procedures to which both products were subjected did not increase the in-vivo rate of plasma disappearance of Factor VIII. Furthermore, the study demonstrated that the pharmacokinetic characteristics of both products were very similar.384
For Factor IX the situation seemed to be quite similar. The stability of different dry heated prothrombin complex concentrates depends on their composition, i.e. on the method of preparation and on additives such as reducing or non-reducing sugars. Alcohol precipitation, for instance, and addition of fructose or sorbitol as stabilisers were unsuitable for products which are intended to be heat-treated in the lyophilised state. Among the tests applied, only clotting-factor activities and, to a minor degree, turbidity, measured at 650 nm, responded significantly to small changes in heat-treatment. A good yield to produce a Factor IX high potency concentrate was found by adsorption on DEAE-Sephadex and ammonium sulphate precipitations. The freeze-dried material could be heated for up to 96 hours and 70 degrees C without any sign of denaturation.385
Using pilot-scale production of a Factor IX (II and X) concentrate, the effects of starting plasma source and processing parameters on two in-vitro indicators of product quality yield and thrombogenic potential were studied. The difference in yield between products derived from cryo-supernatant and Cohn fraction I supernatant was found for all plasma types studied, and was significant (Student’s t-test for paired observations, p<0.001). Heating the products to 60°C for 72 h – the viral sterilisation procedure used – did not affect yield. Heat treatment for 72 h at 80°C, while not affecting product solubility, resulted in an average yield drop of 25% with the difference between products derived from cryo-supernatant and Cohn fraction I supernatant being maintained. This drop in yield was also significant (p<0.001).386
(See also section 9C.1-5)
Heating technology has progressed regarding the equipment (mixing, thermostatic bath, wet autoclave, etc.) in domains such as vaccines and food. It is essential to avoid hot spots in the protein solution by adequate mixing, uniform heat transfer from the tank wall to the solution and throughout the solution, so as to obtain homogeneous warming of the solution. Careful monitoring is also essential. The choice of each item of equipment is thus of high importance. Each step in the different processes should be carefully monitored.
Virus inactivation/removal processes are an additional step that has to be included in the manufacturing plant. For vapour treatment, a special device must be built to inject vapour into closed systems.
For dry heating, the choice of a high-performance freeze-dryer that can also heat the vials is required. The dry-heating process can be carried out separately after the freeze-drying step in a separate oven or autoclave.
It is essential that components that have been subjected to a virus inactivation/removal step are segregated from untreated material.387 The virus inactivation/removal step must be performed in a dedicated area, physically close but separated from the earlier production steps to avoid cross-contamination and recontamination. It requires a complete range of dedicated equipment and qualified staff.
When the treatment is performed at the last production step (e.g. terminal dry-heat), no extra specific material is necessary except an efficient freeze-dryer associated with an oven or an autoclave. No specific area dedicated to this virus inactivation is required.
The monitoring of the virus inactivation step should be rigorous and requires additional specific in-process controls. This virus inactivation by heat is generally an in-process step and is followed by a stabiliser removal step. The application of a viral reduction strategy increases the complexity of the manufacturing process, reduces the production yield to some extent and requires more resources.
For dry heating, a high-performance freeze-dryer that can also heat the vials is required.
Proteins are rapidly denatured when heated. To preserve the activity of a plasma protein during the heating process, carefully selected stabilisers or protective agents such as amino acids, sugars, and polyols should be added to the protein preparation to avoid undesirable denaturation and chemical effects such as decreased solubility, loss of activity, polymerisation, aggregation, and even yellow-coloured Maillard reaction products.388,389 After in-process treatment these stabilisers, often present at high concentration, must be removed, mostly by chromatography or ultrafiltration.
The crucial point in heating PDMPs was finding the best excipient mixture for preserving PDMP thermostability while allowing effective inactivation of the hepatitis B and C viruses and HIV. The approach used was largely experimental in the 1970s-1980s as the knowledge essential to speeding up research was lacking (no theoretical model was available and the structures of plasma proteins were largely unknown). In the 1980s, the methods available for evaluating the safety of PDMPs were extremely limited.
Mostly, virus inactivation treatments were developed within the laboratories of plasma fractionators or by individuals closely associated with these industries.390 These developments were not usually published in scientific journals so that information can only be found in product package inserts.391
The ultimate proof of virus inactivation was the absence of disease in patients. An approach used for HBV and HCV (neither of which could be multiplied in vitro) was evaluated in chimpanzees.392 The chimpanzee model and the conditions used are presented in Prince and Brotman.393
Another technological advance was developments in measuring Factor VIII activity. It should be stressed that the virus inactivation treatments may affect the Factor VIII molecule to some extent and that its activity should be monitored carefully (see section 9B.1.i-iii). Reliable methods for the meaningful quantification of Factor VIII in therapeutic products are essential.394,395 Since the 1970s and 1980s, there have been positive improvements in measuring Factor VIII activity during the production cycle in general and particularly after the virus inactivation steps. In products designed with a specific potency, differences may in part be caused by differences in reagents, procedures, good standards and potentially by the nature of Factor VIII products themselves.
It is recognised that it is difficult to inactivate or remove all known enveloped and non-enveloped viruses with diverse physico-chemical characterisation using a single process step. In addition to virus inactivation by heat, solvent-detergent treatment and nanofiltration are the leading technologies and are often used as one of the virus inactivation steps in the manufacturing processes of plasma-derived products and recombinants.
One of the major advances in human viral safety of PDMPs is solvent/detergent treatment, developed at the New York Blood Center.396,397,398,399
In January 1984, Prince et al published a study demonstrating that 4 concentrates lots of Factor VIII and 2 lots of Factor IX containing titrated stocks of hepatitis B and Hutchinson non-A, non-B hepatitis virus and treated by Tween 80 and 20% ether, did not transmit the infection to two chimpanzees and the animals remained free from infection after 9 months of follow up.400,401
The presence of lipid envelopes on blood-borne viruses such as HIV, HCV, HBV, and SARS-CoV makes them susceptible to inactivation by chemicals that dissolve or dissociate lipids without damaging the constituent proteins. Virologists traditionally used a combination of the detergent Tween 80 (polysorbate 80) and ethyl ether to dissociate lipid-enveloped viruses into subunits. Later, a combination of Tween 80, Triton X100 (octoxynol) or sodium cholate, and the solvent tri(n-butyl) phosphate (TNBP) appeared to work best and was soon applied to a variety of plasma proteins considered at risk of transmitting viruses.402 The rapid and complete destruction of enveloped viruses suggested the safety margin of this methodology. After treatment, the solvent-detergent must be eliminated by one of three methods: oil extraction (solvent-detergent, in contrast to proteins, is soluble in oil), gel filtration chromatography, or ion-exchange chromatography. Solvent/Detergent (SD) is an extraordinarily effective means of eliminating enveloped viruses from plasma. The process is cheap and reliable at industrial scale, its yield is high (more than 90%), and it does not cause protein denaturation.
Further validation studies with the S/D technology were performed in 1985 with HTLV-III (former name for HIV), using plasma products spiked with HIV and the cell line permissive for its growth obtained from Dr R. Gallo as well as model viruses such as VSV, Sindbis virus and Sendai virus in a cellular model.403,404
Clinical safety studies were initiated by the New York Blood Center and the US FDA, Biotransfusion and the French Study Group, Octapharma and the Centro de Hematologica Santa Catharina.
Factor VIII concentrate prepared using this S/D method has been licensed in the US for routine use since June 1985.405 Subsequently, 25 organisations worldwide have adopted the use of S/D in the preparation of PDMPs, and different national authorities have reviewed the technology. Products made by this method are in use in Germany, France, Spain, Italy, Brazil, Israel, Switzerland and Sweden.
However, solvent/detergent is not able to inactivate non-enveloped viruses (no lipid coat). HAV and parvovirus B19 have been transmitted by S/D-treated clotting factors (see section 9B.8.i-iii). Therefore, a second virus inactivation method has been required by the authorities since the 1990s. Solvent-detergent treatment has been applied in the manufacture of Factor IX, Factor VIII, intramuscular immunoglobulin, and intravenous immunoglobulin. It is widely used in the recombinant protein industry. To consolidate their data on virus inactivation by solvent-detergent, seven fractionation companies have collaborated. The results of 308 virus validation studies performed in the laboratory, demonstrate the robustness and the efficacy of the method against enveloped viruses.406 Based on further improvements of the S/D technology, TNBP and Tween 80 are the most used combination today.407
Nanofiltration is a more recent development for virus removal. Effective removal requires that the pore size of the filter is smaller than the effective diameter of the virus.
In the early 1990s, membranes with a multi-layered pore structure appeared, allowing the removal of viruses by size exclusion. These filters had a particularly well controlled pore size distribution in the nanometer (nm) range that allowed the effective removal of virus particles, while passage of the therapeutic proteins was maintained at high yield. The Japanese company Asahi Kasei developed the first available nanofilters, with a pore size of 75 and 35 nm. Later, a range of nanofilters became available with pore sizes of 15 nm, 20 nm, and 50 nm and produced by different manufacturers such as Millipore and Pall.
The choice of membrane to be used depends on the size of the virus and the therapeutic protein. This technology is not easily applied to high molecular weight protein concentrate without significant protein loss using 15 nm membranes. The size of the enveloped viruses HIV, HCV and HBV are respectively 100 nm, 30-60 nm and 42 nm in contrast with the small size (18-30 nm) of the non-enveloped viruses HAV and parvovirus B19. Nanofiltration through 15 nm membranes may eliminate all major viruses including HAV and parvovirus B19 and may eliminate prions. This is not the case using 35 nm membranes.
Nanofiltration was incorporated into the production processes of inhibitors (e.g. antithrombin, A1P1, and C1-inhibitor), immunoglobulin, and coagulation factors such as Factor IX, FVIII, FX, fibrinogen, Factor XIII, plasminogen, and Protein X.408,409,410
A study combining different virus validation results of 7 manufacturers (Bio Products Laboratory, Biotest, CSL Behring, Bern, CSL Behring, Marburg, Grifols, Kedrion, and Takeda) was recently published.411 The studies to validate virus removal using nanofiltration processes were performed using 17 different plasma product intermediates including clotting factors, inhibitors, and immunoglobulins. The results show that the most relevant transfusion-transmitted viruses (HBV, HCV and HIV) with a size from 42 to 100 nm, would be effectively removed during the manufacturing process. The capacity to remove smaller viruses depended on filter pore size, whether the nanofiltration process was integrated and on specific process conditions.
Nanofiltration is a simple, robust, non-destructive process widely used as a second very efficient virus removal step in the production of PDPMs and recombinant proteins. Removal of small viruses can depend on manufacturing process parameters.
Between 1994-1998, treatment at low pH (e.g. pH 4) in the presence or absence of pepsin was originally developed to disaggregate immunoglobulin polymers (see also section 9B.6 and 9B.8).412 The stability of the proteins is sensitive to the degree of protonation of the protein and may unfold to some degree at acidic pH. It is a good technology for inactivating thrombogenic molecules present in an immunoglobulin preparation. This treatment is quite an effective virus inactivation step for enveloped but less effective for non-enveloped viruses.413,414 Virus inactivation at low pH is based on conformational changes in viral structural proteins.415 A typical treatment is pH 4, at 30-37°C, for more than 20h.416,417
Developed in 2003 in addition to fractionation and due to its capacity to induce precipitation in the immunoglobulin manufacturing process, caprylic acid can effectively inactivate enveloped viruses and thrombogenic molecules. The mechanism of inactivation may be interference of the non-ionised form of caprylate and disruption of viral lipid membranes, rendering enveloped viruses non-infectious.418 Monitoring the pH is a crucial factor.419
One manufacturer uses the chemical disinfectant (virucide) sodium thiocyanate as a virus inactivation method (Mononine, CSL Behring).420 It is introduced in an intermediary step in the production of Factor IX.421 Thiocyanate is known as a soft anion, which denatures proteins by inducing protein perturbation to the protein folding.422
Mononine (CSL Behring) or human coagulation Factor IX concentrate, is the first Factor IX purified using monoclonal antibody chromatography.
After development of the product with sodium thiocyanate incubation in 1991, the manufacturing process was approved by the US FDA in 1992. The production was discontinued in 2020.
(See also section 11A).
The downstream processing of essentially all therapeutic proteins is based on similar strategies that combine precipitation, chromatography, ultrafiltration, depth filtration and sterile filtration.
To implement a specific virus inactivation/virus removal method within a process, several points should be considered: regulatory considerations at the time, process consideration and the specific method of virus inactivation chosen. The protein purification process must be considered, not only at the small pilot scale and normal scale, but also at the potential manufacturing scale, to ensure that the virus inactivation/virus removal step is implemented at the best point in the process. Several factors have to be analysed as the protein concentration, impurity concentration, and process volumes may vary throughout the downstream process. Steps such as S/D treatment can be performed earlier in the process where protein concentration may be relatively high; steps such as nanofiltration should be performed where the protein concentration is low. Nevertheless, the protein solution to be treated should be clarified, filtrated and homogenised to assure a good homogeneous protein solution and assure the virus inactivation treatment in good conditions.
For nanofiltration, the choice of filter, the feed for recirculation, the filter capacity, the size of the filter, the pressure and exclusion should be adapted to each therapeutic preparation. Dedicated extra material and validations are required. All these parameters should be carefully defined.
(See section 9C and 10D)
(This section is discussed in section 10E)
Recombinant protein therapeutics were developed to treat a wide variety of clinical disorders, including genetic disorders. They can be produced in bacteria, yeasts or mammalian cells. Only the mammalian cells are capable of producing mammalian proteins with the right glycosylation.
It must be stressed that the production processes of recombinant therapeutics include inactivation steps, as viral infection of mammalian cell culture is a real risk with severe consequences.423
Since 1992, 14 different recombinant coagulation Factor VIII have been approved for haemophilia A treatment by the EMA and FDA. They are full-length FVIII protein, B-domain truncated (Figure 7) or Factor VIII recombinant products with prolonged half-life.424 Since 1997, five different recombinant Factor IX, produced in different animal and human cells, have been approved by the FDA for haemophilia B treatment.
The development of a manufacturing process for a recombinant protein in mammalian cells usually follows a well-established scheme and is well described by Wurm.425
The recombinant gene coding for the protein of interest is first isolated, sequenced and studied. The gene is then linked with the necessary transcriptional elements allowing intracellular protein synthesis, and is transferred into the cells. Murine and human cells are used to produce recombinant Factor VIII.
In addition, a second gene is transferred that confers to recipient cells a selective advantage. In the presence of the selection agent, which is applied a few days after the gene transfer, only those cells that express the selector gene survive (transfection). The most popular genes for selection are dihydrofolate reductase and glutamine synthetase. In both cases, selection occurs in the absence of the appropriate metabolite, preventing the growth of non-transformed cells (transfectants).
Following selection, survivor cells are transferred as single cells to a second cultivation vessel, and the cultures are expanded starting from a single transformed cell (clonal expansion of optimal producer clone). Eventually, the highest cell producers with appropriate growth and production are selected (establishing research, master and working cell banks).
A cultivation process is then established that is determined by the needs (expansion of working cell bank). For recombinant Factor VIII, there are three generations relating to the culture of recombinant cells and the risk of virus contamination during the culture. The two first products, Recombinate and Kogenate (excipient human albumin), are cultivated using animal-derived protein in the culture medium. To avoid animal protein contamination, the second-generation of products are produced in cells multiplying in culture medium containing human proteins. The third-generation involves Factor VIII production in which no animal or human protein is included in the culture medium.
So far, all mammalian recombinant therapeutics are secreted proteins or have been developed from gene constructs that mediate protein secretion in the culture medium. The medium is collected and the recombinant protein is collected and purified.
In more recent clotting factor recombinant generation, no animal or human proteins for cell culture and purification have been used to avoid virus contamination by the culture medium.
The high yield obtained in today’s processes is the result of years of research.
There are two formats for the production of recombinant protein in mammalian cells: cultures of adherent cells in roller flasks and suspension cultures in roller bottles or for large volume, in bio-fermenters. Culture medium is added in a semi-continuous way or by continuously perfused production processes. This latter is used for recombinant FVIII (the largest recombinant produced protein) produced by BHK cells in suspension (Kogenate, Bayer). It is harvested continuously from ongoing perfusion cell cultures. This sophisticated, highly controlled process runs for up to 6 months (see methodology section 12A).
The downstream process of purification, virus inactivation steps, concentration, equipment, monitoring and working in air-controlled rooms are similar to the downstream manufacturing process for PDMPs.
The recombinant FVIII product developed by Genentech is prepared in a continuous cell culture process using baby hamster kidney (BHK) cells transfected with the FVIII gene. Co-expression of recombinant FVIII with von Willebrand Factor in the culture medium is not required for stabilisation of the FVIII molecule, but a special production culture medium containing for example insulin, transferrin and albumin is used.426,427
The Genetics Institute produced recombinant Factor VIII in a batch re-feed cell culture process using Chinese hamster ovary (CHO) cells transfected with the FVIII gene. Co-expression of FVIII with von Willebrand Factor stabilises FVIII. In the absence of von Willebrand Factor, the heavy chain of FVIII expressed in CHO cells is not associated with the light chain and both are degraded. Both recombinant FVIII products are further purified using monoclonal antibody affinity and additional chromatography steps. Since these developments, second and third generation recombinant products have become available.428
Two types of materials and equipment are required for the production of recombinant proteins:
Quality Control with related specific and performing methods and research for protein analysis is required.
A great deal of interest in the possibility of producing ‘synthetic’ FVIII and FIX developed in view of the high rate of transmission of blood borne viruses by plasma-derived concentrates in the 1970s and early 1980s. In 1984, this possibility became reality for FVIII following the successful determination of the structure, cloning and expression of the Factor VIII gene by scientists at the Genetic Institute (Cambridge, MA, US) and Genentech (Berkeley, CA, US). The prospects of viral safety associated with FVIII produced from recombinant DNA technology were the main advantage. Following this development, recombinant FVIII could, at least theoretically, become available in unlimited supply. These achievements were remarkable in view of the size and complexity of the FVIII gene that encompassed 186,000 base pairs and represented 0.1% of the human X chromosome.429,430,431,432
In collaboration with scientists at Genentech and the Genetic Institute, two pharmaceutical companies (Miles, Inc/Cutter Biological, Berkeley, CA, US and Baxter/Hyland Div. Glendale, CA, US) succeeded in developing scale-up, purification and standardisation of two recombinant FVIII products for clinical use. Following preclinical in vitro studies, and studies in animals, pre-license clinical trials in patients with haemophilia A began in 1987.433
Studies on efficacy and safety in the treatment of bleeding episodes and in controlling bleeding in major surgery in adults were then carried out.434,435
Recombinate was licensed for use in the US in 1992 and Kogenate was licensed for use in early 1993. In January 1989, a study in previously untreated patients (PUPs) was begun with Kogenate. One year later, in July 1990, a PUP study with Recombinate started.436,437,438
In the PUP trials, the haemostatic responses were excellent and both products were well tolerated. However, in 20-25% of the study subjects, inhibitors developed early in the treatment (after a median of 9-11 exposure days (EDs)). Approximately 50% of the inhibitors in both PUP studies were ‘high responding’ (>5 Bethesda Units), whereas the remainder were ‘low responding’ and most of these were transient.439,440
For that reason, some clinicians became concerned that recombinant FVIII was causing a higher incidence of inhibitors.
Studies published in 1992 and 1993 on infants and children with severe haemophilia A documented a higher incidence of inhibitor development with plasma-derived FVIII (25-50%) than previously thought.441,442
It has become increasingly apparent that, if inhibitors are studied prospectively, with laboratory monitoring at frequent intervals, 25-35% (or even 50%) of previously untreated patients will be found to develop inhibitors after a median of 9-11 exposure days (EDs). Roughly one-third of these will disappear despite continued exposure to FVIII given for episodic treatment. In addition, it became apparent that such findings were not related to a particular type of product, but were seen with plasma-derived as well as recombinant FVIII products. Other analyses were documenting that patient-related factors, such as the severity of haemophilia A, ethnicity, or a FVIII gene mutation causing the person’s haemophilia, were more important determinants of inhibitor development.
It also became apparent that very few persons with severe haemophilia who received >50 EDs with plasma-derived FVIII developed new inhibitors while on recombinant FVIII. While the benefits of recombinant FVIII products were recognised because of increased viral safety, there was still some concern over the fact that the original recombinant FVIII products, Recombinate and Kogenate, contained pasteurised human serum albumin as stabiliser. Pasteurised human serum albumin had an excellent safety record, and there was no indication that it caused any problems in recipients. Nevertheless, human serum albumin was later replaced by sucrose as stabiliser.443,444
As newer, so-called ‘second generation’ recombinant FVIII products were developed, some clinicians worried that these might be more immunogenic. Pharmacia’s (Stockholm, Sweden) B-domainless recombinant FVIII (rFVIII SQ) entered pre-license clinical trials in 1993 in Europe and in the US in 1995.445,446
No albumin is needed to stabilise B-domainless recombinant FVIII; however, it was used in the manufacture of the product. B-domainless recombinant FVIII (ReFacto, later referred to as Xynthia, Wyeth Pharmaceuticals, Collegeville, PA, US) has not been associated with increased incidence or prevalence of FVIII inhibitors as compared with plasma-derived full length recombinant FVIII products in previously treated patients or previously untreated patients.447
From the introduction of the first recombinant FVIII in the late 1980s, through each new variation, there have been carefully designed, long-term, prospective clinical trials in both previously treated patients and previously untreated patients to assess safety and efficacy. These trials have included frequent inhibitor assays, as well as other laboratory and clinical observations. Each of these recombinant preparations have proven to be safe and effective. None have resulted in an increased incidence or prevalence of inhibitors. On the other hand, a large body of useful information has been gained from these and other studies which have improved our understanding as to which patients are at greater risk of developing an inhibitor, the important genetic and environmental risk factors, and the long term viral safety of FVIII products.448
Patients with haemophilia A who are treated with recombinant Factor VIII sometimes develop antibodies to residual mammalian proteins within the product, and there are reported cases of clinical relevance.449
|
Term |
Definition |
|---|---|
|
Adjuvant |
A substance which enhances the body’s immune response to an antigen. |
|
Adverse event |
An undesired, unintended pharmacological effect that occurs when a medication is administered correctly. |
|
Antibody |
A large, Y-shaped protein used by the immune system to identify and neutralise foreign substances (antigens) such as pathogenic bacteria and viruses, also known as immunoglobulins. |
|
Anticomplementary |
Having the capacity to remove or inactivate complement nonspecifically (see complement action below). |
|
Antigen |
A molecule capable of stimulating an immune response e.g. a protein at the surface of a pathogen. |
|
Apheresis |
A medical procedure that involves removing whole blood from a donor and separating the blood into individual components so that one particular component can be removed. The remaining blood components are then reintroduced back into the bloodstream of the donor. |
|
Caprylic acid |
Also known under the name of octanoic acid, is a saturated fatty acid with the formula CH3(CH2)6C02H. |
|
Caprylic acid treatment |
A process of viral inactivation that has been shown to robustly inactivate or remove infectivity of lipid-enveloped viruses. |
|
Capsid |
Is the protein shell of a virus enclosing its genetic material. |
|
Cell mediated immunity |
An immune response that does not involve antibodies. It is the activation of phagocytes, antigen-specific cytotoxic T-lymphocytes, and the release of various cytokines in response to an antigen. |
|
Chromatography |
A technique for the separation of a mixture of different substances by passing it in solution or suspension through a medium in which the components move at different rates and are separated from each other. |
|
Colloids |
Gelatinous solutions that maintain a high osmotic pressure in the blood. |
|
Complement activation |
Is a cascading event like the falling of a row of dominoes. It must follow a specific order if the end result is to be achieved. The complement system, also known as complement cascade, is a part of the immune system. The complement cascade is a tightly regulated network of plasma proteins and cell surface receptors that recognise non-self-components and triggers one of the three pathways. An antigen-antibody complex triggers the classical pathway; carbohydrates trigger the lectin pathway; and foreign surfaces trigger the alternative pathway. Intermediates of the complement cascade can opsonize cells and peptide byproducts of complement cleavage promote inflammation. |
|
Co-purify |
Where two or more substances (e.g. proteins) attract and form bonds to each other to form a complex such as a protein complex. |
|
Crystalloids |
Isotonic plasma volume expanders that contain electrolytes. |
|
DEAE sephadex |
Diethyl-aminoethyl is a positively-charged slurry that will have electrostatic interactions with the negatively charged atoms and is used as an ion-exchange resin in a process that separates substances based on their charges. |
|
Dry heat |
A method of viral inactivation that involves the heating of protein following lyophilisation. |
|
Dynamic range |
The ratio between the largest and the smallest that a certain quantity (here proteins) can assume. It is often expressed as a logarithmic value. |
|
Endotoxins |
Or lipopolysaccharides, are large molecules consisting of a lipid and a polysaccharide and are found in the outer membrane of Gram-negative bacteria. |
|
Excipient |
An inactive substance that serves as a vehicle or medium for a drug or other active substance. |
|
Glycoforms |
An isoform of a protein that differs only with respect to the number or type of attached glycan. |
|
Haemorrhagic diathesis |
An abnormal tendency to spontaneous severe bleeding. |
|
Heterodimers |
A chemical compound composed of two non-identical subunits. |
|
HLA |
Human Leucocyte Antigen. |
|
Immunogenicity |
The ability of a foreign substance such as an antigen to provoke an immune response. |
|
Inhibitor |
Reduces the activity of a protein, enzyme, hormone or medicinal product. |
|
International standards |
Are used for calibration of assays to measure clotting Factor VIII activity of a concentrate. |
|
International Units |
Factor VIII concentration is expressed in international units (IU); 1 IU is defined as the concentration of coagulation factor in 1 mL of normal pooled plasma. Healthy people have a factor VIII or factor IX plasma concentration of 0.50-1.50 IU/mL. |
|
Isoelectric |
Having or involving no net electric charge or difference in electric potential. |
|
ISTH |
International Society on Thrombosis and Haemostasis. |
|
kDa |
kiloDalton. A unit of mass used to express molecular mass, especially for large molecules, such as proteins and polysaccharides. |
|
Leakage markers |
Prognostic markers in plasma of a change in the normal status of the body. For example, inflammatory serological markers as indicators of complications during the course or after a treatment. |
|
Low-pH incubation |
A process of viral inactivation used to inactivate large enveloped viruses. |
|
Lyophilisation |
Or freeze-drying, is a process used to preserve biological material by freezing and drying the sample under vacuum at very low temperatures, resulting in removal of water from the sample. |
|
Maillard reaction |
Chemical reaction between amino acids and reducing sugars. |
|
Mitogens |
A peptide or small protein that induces a cell to begin cell division: mitosis. |
|
um = μm (micrometre) |
One-millionth of a metre. |
|
Monomeric |
A molecule that can react together with other monomeric molecules to form a larger polymer chain or a three dimensional network in a process called polymerisation. |
|
Nanofiltration |
A process that removes viruses according to their size while permitting flow-through of the desired protein. |
|
Oncotic pressure |
Or colloid osmotic pressure, is a form of osmotic pressure induced by the proteins, notably albumin, in the plasma in a blood vessel that displaces water molecules, thus creating a relative water molecule deficit with water molecules moving back into the circulatory system within the lower venous pressure end of capillaries. |
|
Opsonisation |
Is an immune process which uses opsonins to tag foreign pathogens for elimination by phagocytes. |
|
Orthogonal |
Two different methods based on different principles. |
|
Osmolality |
Is a measure of the electrolytes in water per kg, measured with an osmometer. |
|
Osmolarity |
Is an estimation of the osmolar concentration of a solution and is proportional to the number of particles per litre of solution. |
|
Pasteurisation |
A process through which human plasma is subjected to a viral inactivation treatment by heating it in solution (the liquid state) for 10 hours at 60° C (from Pasteur's methodology). |
|
PDMP |
Plasma derived medicinal product |
|
Physicochemical |
Being physical and chemical. |
|
Plasma proteome |
The entire set of proteins that are or can be expressed from the human genome. |
|
Plasmapheresis |
A medical procedure that involves removing whole blood from a donor and separating the blood into individual components so that one particular component can be removed. The remaining blood components are then reintroduced back into the bloodstream of the donor. |
|
Polyvalent |
Active against several antigens, in the case of immunoglobulins. |
|
PPSB |
A plasma derived medicinal product that contains the blood clotting factors Prothrombin (Factor II), Proconvertin (Factor VII), Stuart-Prower factor (Factor X) and anti-haemophilic factor B (Factor IX). |
|
Precipitate |
Sediment or deposit. |
|
Prekallikrein |
Is a serine protease that complexes with high-molecular-weight kininogen. |
|
Protease |
An enzyme that catalyses (increases the rate of) proteolysis, the breakdown of proteins into smaller polypeptides or single amino acids. |
|
Proteome |
The proteome is the entire set of proteins, glycosylated or not, that is or can be expressed by a cell, tissue, or an organism. |
|
Purpura fulminans |
An acute purpuric rash characterised by coagulation of the micro vasculature. |
|
Pyrogenic |
Fever-inducing. |
|
Pyrogenicity |
Capacity to produce fever. |
|
RNA |
Ribonucleic acid (RNA) is involved in translation and transcription, which are the mechanisms by which cells express genes. |
|
Seroconversion |
The transition from infection with a virus (such as HIV) or other pathogen to the detectable presence of pathogen-specific antibodies in the blood. |
|
Side effect |
An effect, whether therapeutic or adverse, that is secondary to the intended one. The term is predominantly employed to describe unwanted adverse effects. |
|
S-sulfonation |
Chemical reaction transferring an –SO3 group to an alkane or aromatic group. |
|
Solvent/detergent |
A process using a mixture of detergent in the presence of a solvent that inactivates enveloped viruses in pooled plasma and in plasma-derived-medicinal products virtually eliminates the risk of transmission of enveloped viruses. |
|
Thrombogenicity |
Tendency to generate blood clotting and/or thrombosis. |
|
Tolerogenic: |
Capable of producing immunological tolerance. |
|
TNBP |
Tri(n-butyl) phosphate is a solvent that when combined with a detergent, Tween 80 or Triton X100, inactivates efficiently enveloped viruses. |
|
TSE Task Force |
Transmissible Spongiform Encephalopathy Task Force. |
|
Wet heat |
The product is saturated with water vapour (high level of humidity), but it is not in solution. |
1 Anderson, N.L., & Anderson, N.G. (2002). “The Human Plasma Proteome: history, character, and diagnostic prospects”. Molecular & Cellular Proteomics, 1(11), 845-867.
2 Cohn E.J. et al. (1944). “Chemical, Clinical, and Immunological Studies on the Products of Human Plasma Fractionation: The Characterization of the Protein Fractions of Human Plasma”. Journal of Clinical Investigation, 23(4), 417-432.
3 Ala, F, Burnouf, T., El-Nageh, M. (1999). Plasma fractionation programmes for developing countries: technical aspects and infrastructural requirements. Alexandria, Egypt: WHO, Regional Office for the Eastern Mediterranean. [online] Available at: https://apps.who.int/iris/handle/10665/119608.
4 Curling, J.M. (2002). “International New Technology into Blood Plasma Fractionation”. BioPharm International, 15(9), 16-26. [online] Available at: https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.562.8430&rep=rep1&type=pdf.
5 Grabowski, H., Manning, R. (2018, February). Key economic and value considerations in the US market for plasma protein therapies. Bates White. https://www.bateswhite.com/media/publication/154_Plasma%20Protein%20Therapies%20paper.pdf.
6 Ala, F, Burnouf, T., El-Nageh, M. (1999). Plasma fractionation programmes for developing countries: technical aspects and infrastructural requirements. Alexandria, Egypt: WHO, Regional Office for the Eastern Mediterranean. [online] Available at: https://apps.who.int/iris/handle/10665/119608.
7 Curling, J.M. (2002). “International New Technology into Blood Plasma Fractionation”. BioPharm International, 15(9), 16-26. [online] Available at: https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.562.8430&rep=rep1&type=pdf.
8 Grabowski, H., Manning, R. (2018, February). Key economic and value considerations in the US market for plasma protein therapies. Bates White. https://www.bateswhite.com/media/publication/154_Plasma%20Protein%20Therapies%20paper.pdf.
9 Regulation 141/2000 of the European Parliament and of the Council of 16 December 1999 on orphan medicinal products. (2000). OJ, (L18), 1–5.
10 NIH.gov. (2017). FAQs About Rare Diseases | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. [online] Available at: https://rarediseases.info.nih.gov/diseases/pages/31/faqs-about-rare-diseases.
11 World Health Organization. (2021). Guidance on increasing supplies of plasma-derived medicinal products in low- and middle-income countries through fractionation of domestic plasma. [online] Available at: https://www.who.int/publications/i/item/9789240021815.
12 World Health Organization. (2019). World Health Organization Model List of Essential Medicines: 21st List. [online] Available at: https://apps.who.int/iris/handle/10665/325771.
13 Burnouf, T. et al. (2020). “Plasma fractionation in countries with limited infrastructure and low-/medium income: How to move forward?”. Transfusion and Apheresis Science, 59(1). [online] Available at: https://doi.org/10.1016/j.transci.2019.102715.
14 Strengers, P.F.W. (2017). “Evidence-based clinical indications of plasma products and future prospects”. Annals of Blood, 2(20), 1-7.
15 Quinlan, G.J., Martin, G.S., & Evans, T.W. (2005). “Albumin: Biochemical Properties and Therapeutic Potential”. Hepatology, 41(6), 1211-1219.
16 World Health Organization. (no date). Biologicals. [online] Available at: https://www.who.int/health-topics/biologicals#tab=tab_1.
17 Working Together for a Safe and Sustainable Blood Supply. (no date). [online] Available at: https://www.edqm.eu/sites/default/files/medias/fichiers/Transfusion/Blood_Guide/10_years_ec-edqm_co-operation_in_the_field_of_blood.pdf.
18 Gov.uk. (no date). Quality and safety of human blood and blood products. [online] Available at: https://www.gov.uk/guidance/quality-and-safety-of-human-blood-and-blood-products.
19 Directive 2002/98/EC of the European Parliament and of the Council of 27 January 2003 setting standards of quality and safety for the collection, testing, processing, storage and distribution of human blood and blood components and amending Directive 2001/83/EC. (2003). OJ, (L33), 30-40.
20 Commission Directive 2004/33/EC of 22 March 2004 implementing Directive 2002/98/EC of the European Parliament and of the Council as regards certain technical requirements for blood and blood components. (2004). OJ, (L91), 25-39.
21 Commission Directive 2005/61/EC of 30 September 2005 implementing Directive 2002/98/EC of the European Parliament and of the Council as regards traceability requirements and notification of serious adverse reactions and events. (2006). OJ (L287M), 350-358.
22 Commission Directive 2005/62/EC of 30 September 2005 implementing Directive 2002/98/EC of the European Parliament and of the Council as regards Community standards and specifications relating to a quality system for blood establishments. (2005). OJ, (L256). 41-48.
23 Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. (2001). OJ, (L311), 67-128.
24 European Commission. (no date). Good Manufacturing Practice – Volume 4 of ‘The rules governing medicinal products in the European Union’. [online] Available at: https://ec.europa.eu/health/documents/eudralex/vol-4_en.
25 World Health Organization. (2009). Screening donated blood for transfusion-transmissible infections: recommendations. [online] Available at: https://apps.who.int/iris/bitstream/handle/10665/44202/9789241547888_eng.pdf?sequence=1&isAllowed=y.
26 Chandler, T. et al. (2021). “Blood donation in times of crisis: Early insight into the impact of Covid 19 on blood donors and their motivation to donate across European countries”. Vox Sanguinis. [online] Available at: https://doi.org/10.1111/vox.13103.
27 Watt, J.G. et al. (1972). “New Developments in Large-scale Plasma Fractionation”. Proceedings of the Royal Society of Edinburgh, Section B: Biological Sciences. 71(3), 15-34.
28 Curling, J.M. (1982, August 1-7). Current practice and future possibilities in plasma protein fractionation. Joint meetings of the 19th Congress of the International Society of Haematology and the 17th Congress of the International Society of Blood Transfusion: Hungary, Budapest.
29 European Committee (Partial Agreement) on Blood Transfusion (CD-P-TS). (2020). Guide to the preparation, use and quality assurance of blood components. EDQM: 20th Edition.
30 The European Medicines Agency, Committee for medicinal products for human use. (2011). “Guideline on Plasma-Derived Medicinal Products”, EMA/CHMP/BWP/706271/2010. [online] Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-plasma-derived-medicinal-products_en.pdf.
31 World Health Organization. (2021). Guidance on increasing supplies of plasma-derived medicinal products in low- and middle-income countries through fractionation of domestic plasma. [online] Available at: https://www.who.int/publications/i/item/9789240021815.
32 Hellstern, P. et al. (2001). “The impact of the intensity of serial automated plasmapheresis and the speed of deep-freezing on the quality of plasma”. Transfusion, 41(12), 1601-1605.
33 Laub, R. et al. (2010). “Specific protein content of pools of plasma for fractionation from different sources: impact of frequency of donations”. Vox Sanguinis, 99(3), 220-231.
34 Burkhardt, T., Rothe, R., & Moog, R. (2017). “Immunoglobulin G levels during collection of large volume plasma for fractionation”. Transfusion and Apheresis Science, 56(3), 417-420.
35 Prevot, J., & Jolles, S. (2020). “Global immunoglobulin supply: steaming towards the iceberg?”. Current Opinion in Allergy and Clinical Immunology, 20(6), 557-564.
36 Schreiber, G.B., & Kimber, M.C. (2017). “Source Plasma Donors: A Snapshot”. Transfusion, 57(S3), 110A.
37 Prevot, J., & Jolles, S. (2020). “Global immunoglobulin supply: steaming towards the iceberg?”. Current Opinion in Allergy and Clinical Immunology, 20(6), 557-564.
38 Van der Poel, C. L., Seifried, E. & Schaasberg, W.P. (2002). “Paying for blood donations: still a risk?”. Vox Sanguinis, 83(4), 285-93.
39 Van der Poel, C.L., Seifried, E., & Schaasberg, W.P. (2002). “Paying for blood donations: still a risk?” Vox Sanguinis, 83(4), 285-293.
40 The European Agency for the Evaluation of Medicinal Products, Committee for Proprietary Medicinal Products. (2001). “Non-Remunerated and Remunerated Donors: Safety and Supply of Plasma-Derived Medicinal Products”. EMEA/CPMP/BWP/1818/02/Final, [online] Available at: https://www.ema.europa.eu/en/documents/position/cpmp-position-statement-non-remunerated-remunerated-donors-safety-suply-plasma-derived-medicinal_en.pdf.
41 Hartman. J., & Klein, H.G. (2020). “Supply and demand for plasma-derived medicinal products – A critical reassessment amid the COVID-19 pandemic”. Transfusion, 60(11), 2748-2752.
42 World Health Organization. (2021). Guidance on increasing supplies of plasma-derived medicinal products in low- and middle-income countries through fractionation of domestic plasma. [online] Available at: https://www.who.int/publications/i/item/9789240021815.
43 NHS Blood Donation. (no date). [online] Available at: http://blood.co.uk.
44 World Health Organization. (2021). Guidance on increasing supplies of plasma-derived medicinal products in low- and middle-income countries through fractionation of domestic plasma. [online] Available at: https://www.who.int/publications/i/item/9789240021815.
45 World Health Organization. (2009). Screening donated blood for transfusion-transmissible infections: recommendations. [online] Available at: https://apps.who.int/iris/bitstream/handle/10665/44202/9789241547888_eng.pdf.
46 National Institute for Biological Standards and Control. (no date). Plasma pool testing. [online] Available at: http://www.nibsc.org/science_and_research/idd/plasma_pool_testing.aspx/.
47 The European Medicines Agency, Committee for Medicinal Products for Human Use. (2006). “Guideline on the Scientific Data Requirements for a Plasma Master File (PMF)”, EMEA/CHMP/BWP/3794/03 rev.1. [online] Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-scientific-data-requirements-plasma-master-file-pmf-revision-1_en.pdf.
48 The European Agency for the Evaluation of Medicinal Products, Committee for Proprietary Medicinal Products. (2004). “Guideline on Requirements For Plasma Master File (PMF) Certification”, EMEA/CPMP/BWP/4663/03.
[online] Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-requirements-plasma-master-file-pmf-certification_en.pdf.
49 Council of Europe. (2007). European Pharmacopoeia 6.2. monograph (07/2008:0853), “Anti-A and anti-B haemagglutinins (2.6.20), Anti-D antibodies (2.6.26)”.
50 The European Medicines Agency, Committee for Medicinal Products for Human Use. (2006). “Guideline on the Scientific Data Requirements for a Plasma Master File (PMF)”, EMEA/CHMP/BWP/3794/03 rev.1. [online] Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-scientific-data-requirements-plasma-master-file-pmf-revision-1_en.pdf.
51 Cohn, E.J. et al. (1946). “Preparation and Properties of Serum and Plasma Proteins. IV. A System for the Separation into Fractions of the Protein and Lipoprotein Components of Biological Tissues and Fluids”. Journal of the American Chemical Society, 68(3), 459-475.
52 Edsall, J.T. (1961). “Edwin Joseph Cohn 1892-1953 A Biographical Memoir”. National Academy of Sciences. Washington, D.C. [online] Available at: http://www.nasonline.org/publications/biographical-memoirs/memoir-pdfs/cohn-edwin-j.pdf
53 Van Alstine, J.M., Jagschies, G., & Lacki, K.M. (2018). Alternative Separation Methods: Flocculation and Precipitation. In G. Jagschies, E. Lindskog, K. Łącki, P. Galliher (Eds.), Biopharmaceutical Processing. Elsevier, 221-239.
54 Lynch T.J. et al. (1996). “Considerations of pool size in the manufacture of plasma derivatives”. Transfusion, 36(9), 70-775.
55 Warshel, A. (1987). “What about protein polarity?”. Nature, 330, 15-16.
56 Cohn, E.J. et al. (1946). “Preparation and Properties of Serum and Plasma Proteins. IV. A System for the Separation into Fractions of the Protein and Lipoprotein Components of Biological Tissues and Fluids”. Journal of the American Chemical Society, 68(3), 459-475.
57 Edsall, J.T. (1961). “Edwin Joseph Cohn 1892-1953 A Biographical Memoir”. National Academy of Sciences. Washington, D.C. [online] Available at: http://www.nasonline.org/publications/biographical-memoirs/memoir-pdfs/cohn-edwin-j.pdf.
58 Cohn, E.J. et al. (1946). “Preparation and Properties of Serum and Plasma Proteins. IV. A System for the Separation into Fractions of the Protein and Lipoprotein Components of Biological Tissues and Fluids”. Journal of the American Chemical Society, 68(3), 459-475.
59 Kendrick, D.B. et al. (1964). “Blood program in World War II”. Office of the Surgeon General, Department of the Army. [online] Available at: https://collections.nlm.nih.gov/catalog/nlm:nlmuid-0014773-bk.
60 Schneider, W. et al. (1975). “An alternative method of large scale plasma fractionation for the isolation of serum albumin”. Blut Zeitschrift für die Gesamte Blutforschung , 30(2), 121-134.
61 Schmidt, P.J. (2012). “The plasma wars: a history”. Transfusion, 52, 2S-4S.
62 Creager, A.N.H. (2001). “From blood fractions to antibody structure: gamma globulin research growing out of World War II”. In A.M. Moulin & A. Cambrosio (eds.). Singular Selves. Historical Issues and Contemporary Debates in Immunology. Elsevier.
63 Kendrick, D.B. et al. (1964). “Blood program in World War II”. Office of the Surgeon General, Department of the Army. [online] Available at: https://collections.nlm.nih.gov/catalog/nlm:nlmuid-0014773-bk.
64 Creager, A.N.H. (1999). “‘What blood told Dr Cohn’: World War II, Plasma Fractionation, and the Growth of Human Blood Research”. Studies in History and Philosophy of Bological and Biomedical Sciences, 30(3), 377-405.
65 Cohn, E.J. et al. (1946). “Preparation and Properties of Serum and Plasma Proteins. IV. A System for the Separation into Fractions of the Protein and Lipoprotein Components of Biological Tissues and Fluids”. Journal of the American Chemical Society, 68(3), 459-475.
66 Kendrick, D.B. et al. (1964). “Blood program in World War II”. Office of the Surgeon General, Department of the Army. [online] Available at: https://collections.nlm.nih.gov/catalog/nlm:nlmuid-0014773-bk.
67 Creager, A.N.H. (2001). “From blood fractions to antibody structure: gamma globulin research growing out of World War II”. In A.M. Moulin & A. Cambrosio (eds.). Singular Selves. Historical Issues and Contemporary Debates in Immunology. Elsevier.
68 Creager, A.N.H. (1999). “‘What blood told Dr Cohn’: World War II, Plasma Fractionation, and the Growth of Human Blood Research”. Studies in History and Philosophy of Bological and Biomedical Sciences, 30(3), 377-405.
69 Cohn, E.J. (1945). “Blood Proteins and Their Therapeutic Value”. Science, 101(2612), 51-56.
70 Cohn, E.J. et al. (1946). “Preparation and Properties of Serum and Plasma Proteins. IV. A System for the Separation into Fractions of the Protein and Lipoprotein Components of Biological Tissues and Fluids”. Journal of the American Chemical Society, 68(3), 459-475.
71 Kendrick, D.B. et al. (1964). “Blood program in World War II”. Office of the Surgeon General, Department of the Army. [online] Available at: https://collections.nlm.nih.gov/catalog/nlm:nlmuid-0014773-bk.
72 Ala, F, Burnouf, T., El-Nageh, M. (1999). Plasma fractionation programmes for developing countries: technical aspects and infrastructural requirements. Alexandria, Egypt: WHO, Regional Office for the Eastern Mediterranean. [online] Available at: https://apps.who.int/iris/handle/10665/119608.
73 Watt, J.G. et al. (1972). “New Developments in Large-scale Plasma Fractionation”. Proceedings of the Royal Society of Edinburgh, Section B: Biological Sciences. 71(3), 15-34.
74 Lynch T.J. et al. (1996). “Considerations of pool size in the manufacture of plasma derivatives”. Transfusion, 36(9), 70-775.
75 Oncley, J.L. et al. (1949). “The separation of the antibodies, isoagglutinins, prothrombin, plasminogen and beta1-lipoprotein into subfractions of human plasma”. Journal of the American Chemical Society, 71(2), 541-550.
76 Kistler, P. & Nitschmann H. (1962). “Large scale production of human plasma fractions – eight year experience with the alcohol fractionation procedure of Nitschmann, Kistler and Lergier”. Vox Sanguinis, 7, 414-424.
77 Watt, J.G. et al. (1972). “New Developments in Large-scale Plasma Fractionation”. Proceedings of the Royal Society of Edinburgh, Section B: Biological Sciences. 71(3), 15-34.
78 Cohn, E.J. et al. (1946). “Preparation and Properties of Serum and Plasma Proteins. IV. A System for the Separation into Fractions of the Protein and Lipoprotein Components of Biological Tissues and Fluids”. Journal of the American Chemical Society, 68(3), 459-475.
79 Oncley, J.L. et al. (1949). “The separation of the antibodies, isoagglutinins, prothrombin, plasminogen and beta1-lipoprotein into subfractions of human plasma”. Journal of the American Chemical Society, 71(2), 541-550.
80 Kistler, P. & Nitschmann H. (1962). “Large scale production of human plasma fractions – eight year experience with the alcohol fractionation procedure of Nitschmann, Kistler and Lergier”. Vox Sanguinis, 7, 414-424.
81 Berntorp, E. (1997). Factor VIII Concentrates. In: C. D. Forbes, L. Aledort & R. Madhok (Eds.), Hemophilia. London, New York: Chapman & Hall, 181-192.
82 Nordic Hemophilia Council guideline working group. (2015). [online] Available at: www.nordhemophilia.org.
83 Fasano, M. et al. (2005). “The extraordinary ligand binding properties of human serum albumin”. IUBMB life, 57(12), 787–796.
84 Cohn, E.J. (1945). “Blood Proteins and Their Therapeutic Value”. Science, 101(2612), 51-56.
85 Surgenor, D.M. (2002)., Edwin J. Cohn and the development of protein chemistry. Center for Blood Research Inc. Harvard University Press.
86 Watt, J.G. et al. (1972). “New Developments in Large-scale Plasma Fractionation”. Proceedings of the Royal Society of Edinburgh, Section B: Biological Sciences. 71(3), 15-34.
87 Ala, F, Burnouf, T., El-Nageh, M. (1999). Plasma fractionation programmes for developing countries: technical aspects and infrastructural requirements. Alexandria, Egypt: WHO, Regional Office for the Eastern Mediterranean. [online] Available at: https://apps.who.int/iris/handle/10665/119608.
88 Friedli, H. et al. (1976). “Studies on New Process Procedures in Plasma Fractionation on an Industrial Scale”. Vox Sanguinis, 31(4), 289-295.
89 Matejtschuk, P., Dash, C.H. & Gascoigne, E.W. (2000). “Production of human albumin solution: a continually developing colloid”. British journal of anaesthesia, 85(6), 887-95.
90 Gellis, S.S. et al. (1945). “The use of human immune serum globulin gamma globulin in infectious (epidemie) Hepatitis in the Mediterranean Theater of Operations I. Studies on prophylaxis in two epidemics of infectious Hepatitis”. Journal of the American Medical Association, 128(15), 1062-1063.
91 Dichtelmüller, H.O. et al. (2011). “Contribution to safety of immunoglobulin and albumin from virus partitioning and inactivation by cold ethanol fractionation: a data collection from Plasma Protein Therapeutics Association member companies”. Transfusion, 51(7), 1412-1430.
92 Gellis, S.S. et al. (1945). “The use of human immune serum globulin gamma globulin in infectious (epidemie) Hepatitis in the Mediterranean Theater of Operations I. Studies on prophylaxis in two epidemics of infectious Hepatitis”. Journal of the American Medical Association, 128(15), 1062-1063.
93 Schneider, W. et al. (1975). “An alternative method of large scale plasma fractionation for the isolation of serum albumin”. Blut Zeitschrift für die Gesamte Blutforschung , 30(2), 121-134.
94 Cohn, E.J. (1945). “Blood Proteins and Their Therapeutic Value”. Science, 101(2612), 51-56.
95 Gellis, S.S. et al. (1945). “The use of human immune serum globulin gamma globulin in infectious (epidemie) Hepatitis in the Mediterranean Theater of Operations I. Studies on prophylaxis in two epidemics of infectious Hepatitis”. Journal of the American Medical Association, 128(15), 1062-1063.
96 Hollinger, F. B. (1990). Hepatitis B Virus. In: B. N. Fields & D. M. Knipe (Eds.), Fields Virology. New York: Raven Press, 2171-2236.
97 Geiger, H., Kranz, T., & Haupt, H. (1973). “The Fate of Australia Antigen During Plasma Fractionation”. Developments in Biological Standardization, 27, 158-165.
98 Gellis, S.S. et al. (1945). “The use of human immune serum globulin gamma globulin in infectious (epidemie) Hepatitis in the Mediterranean Theater of Operations I. Studies on prophylaxis in two epidemics of infectious Hepatitis”. Journal of the American Medical Association, 128(15), 1062-1063.
99 The European Agency for the Evaluation of Medicinal Products, Committee for Proprietary Medicinal Products. (1996). “Note for guidance on virus validation studies: the design, contribution and interpretation of studies validating the inactivation and removal of viruses”, CPMP/BWP/268/95. [online] Available at: http://wwweurope.eu/docs/en_GB/document_library/Scientific_guideline wc 50000 3684.pdf2009/09/.
100 Dichtelmüller, H.O. et al. (2011). “Contribution to safety of immunoglobulin and albumin from virus partitioning and inactivation by cold ethanol fractionation: a data collection from Plasma Protein Therapeutics Association member companies”. Transfusion, 51(7), 1412-1430.
101 Curling, J.M. (1982, August 1-7). Current practice and future possibilities in plasma protein fractionation. Joint meetings of the 19th Congress of the International Society of Haematology and the 17th Congress of the International Society of Blood Transfusion: Hungary, Budapest.
102 Ala, F.A. (1992). Frozen fresh plasma and albumin consumption in the United Kingdom. In: U. Rossi, W.G. van Aken & M. Orlando (Eds.), Therapy with plasma and albumin: production and clinical use SIITS-AICT, 17-29.
103 Petersen, I.P., & Bird, A.R. (2004). Small pool heat-treated intermediate purity Factor VIII concentrate. World Federation of Hemophilia. [online] Available at: http://www1.wfh.org/publication/files/pdf-1223.pdf.
104 Zuckerman, A.J. et al. (1971). “The Australia (Hepatitis-associated) antigen in fibrinogen and other fractions of human plasma”. Journal of Clinical Pathology, 24(1), 2-7.
105 Schneider, W. et al. (1975). “An alternative method of large scale plasma fractionation for the isolation of serum albumin”. Blut Zeitschrift für die Gesamte Blutforschung , 30(2), 121-134.
106 Pool, J.G., Hershgold, E.J. & Pappenhagen, A.R. (1964). “High-potency antihaemophilic factor concentrate prepared from cryoglobulin precipitate”. Nature, 203(4942), 312.
107 Pool, J.G. & Shannon, A.E. (1965). “Production of high-potency concentrates of antihemophilic globulin in a closed-bag system”. The New England Journal of Medicine, 273(27), 1443-1447.
108 Smit Sibinga, C.Th. (1986). “Small-pool high yield Factor VIII production”. CRC Critical Reviews in Clinical Laboratory Sciences, 24, 43-70.
109 Watt, J.G. et al. (1972). “New Developments in Large-scale Plasma Fractionation”. Proceedings of the Royal Society of Edinburgh, Section B: Biological Sciences. 71(3), 15-34.
110 Petersen, I.P., & Bird, A.R. (2004). Small pool heat-treated intermediate purity Factor VIII concentrate. World Federation of Hemophilia. [online] Available at: http://www1.wfh.org/publication/files/pdf-1223.pdf.
111 Aledort, L.M. (1998). The treatment of hemophilia: comprehensive care. In: C.D. Forbes, L. Aledort & R. Madhok (Eds.), Hemophilia. London, New York: Chapman & Hall.
112 Edelman, G. & Porter, R. (2006). B Cell Receptor-Structure and Effector Function. In: W. Mak & M.E. Saunders (Eds.), The Immune Response: Basic and Clinical Principles. Elsevier, 93-120.
113 World Health Organization Expert Committee. (1966). The use of human immunoglobulin: Report of a WHO Expert Committee (Technical Report Series Number 327). World Health Organization. [online] Available at: https://apps.who.int/iris/handle/10665/39831.
114 Eibl, M.M. (2008). “History of immunoglobulin replacement”. Immunology and allergy clinics of North America, 28(4), 737-764.
115 Watt, J.G. et al. (1972). “New Developments in Large-scale Plasma Fractionation”. Proceedings of the Royal Society of Edinburgh, Section B: Biological Sciences. 71(3), 15-34.
116 Serra, A. et al. (2021). “Characterization of antibodies in human immunoglobulin products from different regions worldwide”. International Journal of Infectious Diseases, 104, 610-616.
117 Hooper, J.A. (2008). “Intravenous Immunoglobulins: Evolution of Commercial IVIG Preparations”. Immunology and allergy clinics of North America, 28, 765-778.
118 Gelfand, E.W. (no date). Selecting Appropriate IGIV Therapy. MedScape. [online] Available at: http://www.medscape.org/viewarticle/461288. [Accessed 22 April 2021].
119 Farrugia, A., Cassar, J. (2012). “Plasma-derived-medicines: access and usage issues”. Blood transfusion, 10(3), 273-278.
120 Schroeder, H.W. Jr., & Dougherty, C.J. (2012). “Review of intravenous immunoglobulin replacement therapy trials for primary humoral immunodeficiency patients”. Infection, 40(6), 601-611.
121 Bruton, O.C. (1952). “Agammaglobulinemia”. Pediatrics, 9(6), 722-728.
122 Misbah, S. et al. (2009). “Subcutaneous immunoglobulin: opportunities and outlook”. Clinical and experimental immunology, 158(1), 51-59.
123 Barahona Afonso, A.F. & Pires Joao, C.M. (2016). “The Production Processes and Biological Effects of Intravenous Immunoglobulin”. Biomolecules, 6(15), 1-20.
124 Schroeder, H.W. Jr., & Dougherty, C.J. (2012). “Review of intravenous immunoglobulin replacement therapy trials for primary humoral immunodeficiency patients”. Infection, 40(6), 601-611.
125 Hill, L.E. & Mollison, P.L. (1971). Hypogammaglobulinaemia in the United Kingdom. Conclusions. Special Report Series (Medical Research Council (Great Britain)), 310.
126 Eibl, M.M. (2008). “History of immunoglobulin replacement”. Immunology and allergy clinics of North America, 28(4), 737-764.
127 Imbach, P. et al. (1981). “High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood”. Lancet, 1(8232), 1228–1231.
128 Buchacher, A. & Iberer, G. (2006). “Purification of intravenous immunoglobulin G from human plasma-aspects of yield and virus safety”. Biotechnology Journal, 1(2), 148-163.
129 Buchacher, A. & Curling, J.M. (2018). “Alternative Separation Methods: Flocculation and Precipitation”. In G. Jagschies, E. Lindskog, K. Lacki, P. Galliher (Eds.), Biopharmaceutical Processing. Elsevier, 857-876.
130 Oncley, J.L. et al. (1949). “The separation of the antibodies, isoagglutinins, prothrombin, plasminogen and beta1-lipoprotein into subfractions of human plasma”. Journal of the American Chemical Society, 71(2), 541-550.
131 Schneider, W. et al. (1975). “An alternative method of large scale plasma fractionation for the isolation of serum albumin”. Blut Zeitschrift für die Gesamte Blutforschung , 30(2), 121-134.
132 Kistler, P. & Nitschmann H. (1962). “Large scale production of human plasma fractions – eight year experience with the alcohol fractionation procedure of Nitschmann, Kistler and Lergier”. Vox Sanguinis, 7, 414-424.
133 Van Alstine, J.M., Jagschies, G., & Lacki, K.M. (2018). Alternative Separation Methods: Flocculation and Precipitation. In G. Jagschies, E. Lindskog, K. Łącki, P. Galliher (Eds.), Biopharmaceutical Processing. Elsevier, 221-239.
134 Barahona Afonso, A.F. & Pires Joao, C.M. (2016). “The Production Processes and Biological Effects of Intravenous Immunoglobulin”. Biomolecules, 6(15), 1-20.
135 Radosevich, M. & Burnouf, T. (2010). “Intravenous immunoglobulin G: trends in production methods, quality control and quality assurance”. Vox Sanguinis, 98(1), 12-28.
136 Hooper, J.A. (2008). “Intravenous Immunoglobulins: Evolution of Commercial IVIG Preparations”. Immunology and Allergy Clinics of North America, 28, 765-778.
137 Gelfand, E.W. (no date). Selecting Appropriate IGIV Therapy. MedScape. [online] Available at: http://www.medscape.org/viewarticle/461288.
138 Schroeder, H.W. Jr., & Dougherty, C.J. (2012). “Review of intravenous immunoglobulin replacement therapy trials for primary humoral immunodeficiency patients”. Infection, 40(6), 601-611.
139 Späth, P. J. (2017). Essentials of the Production of Safe and Efficacious State-of-the-Art Polyclonal IgG Concentrates. In: P. Imbach (Eds.), Antibody Therapy. Springer, 151-173. .[online] Available at: https://doi.org/10.1007/978-3-319-68038-5_12.
140 Lebing, W. et al. (2003). “Properties of a new intravenous immunoglobulin (IGIV-C,10%) produced by virus inactivation with caprylate and column chromatography”. Vox Sanguinis, 84(3), 193-2001.
141 Hoefferer, L. et al. (2015). “Isoagglutinin reduction by a dedicated immunoaffinity chromatography step in the manufacturing process of human immunoglobulin products”. Transfusion, 55(S2), S117–S121.
142 Eibl, J. et al. (1988). “Ways to reduce the risk of transmission of viral infections by plasma and plasma products. A comparison of methods, their advantages and disadvantages”. Vox Sanguinis, 54(4), 228–245.
143 Ammann, E. M. et al. (2016). “Intravenous immune globulin and thromboembolic adverse events in patients with hematologic malignancy.” Blood, 127(2), 200–207.
144 Daniel, G. W. et al. (2012). “Immune globulins and thrombotic adverse events as recorded in a large administrative database in 2008 through 2010”. Transfusion, 52(10), 2113–2121.
145 Roifman, C. M. et al. (2003). “Comparison of the efficacy of IGIV-C, 10% (caprylate/chromatography) and IGIV-SD, 10% as replacement therapy in primary immune deficiency. A randomized double-blind trial”. International Immunopharmacology, 3(9), 1325–1333.
146 Vo, A. A. et al. (2006). “Safety and adverse events profiles of intravenous gammaglobulin products used for immunomodulation: a single-center experience”. Clinical Journal of the American Society of Nephrology: CJASN, 1(4), 844–852.
147 Dhainaut, F. et al. (2013). “In vitro and in vivo properties differ among liquid intravenous immunoglobulin preparations.” Vox Sanguinis, 104(2), 115–126.
148 Lemm, G. (2002). “Composition and properties of IVIg preparations that affect tolerability and therapeutic efficacy”, Neurology, 59(12) (Suppl 6), S28-S32.
149 Gelfand, E.W. (2005). “Critical Decisions in Selecting an Intravenous Immunoglobulin Product”. Journal of Infusion Nursing, 28(6), 366-374.
150 Gelfand, E.W. (2001). “Antibody-directed therapy: Past, present and future”. Journal of Allergy and Clinical Immunolology, 108(4S), S111-S116.
151 Gelfand, E.W. (no date). Selecting Appropriate IGIV Therapy. MedScape. [online] Available at: http://www.medscape.org/viewarticle/461288.
152 Vo, A. A. et al. (2006). “Safety and adverse events profiles of intravenous gammaglobulin products used for immunomodulation: a single-center experience”. Clinical journal of the American Society of Nephrology: CJASN, 1(4), 844–852.
153 Barahona Afonso, A.F. & Pires Joao, C.M. (2016). “The Production Processes and Biological Effects of Intravenous Immunoglobulin”. Biomolecules, 6(15), 1-20.
154 Daniel, G. W. et al. (2012). “Immune globulins and thrombotic adverse events as recorded in a large administrative database in 2008 through 2010”. Transfusion, 52(10), 2113–2121.
155 Barsoum, N. R. et al. (2002). “Treatment of parvovirus B-19 (PV B-19) infection allows for successful kidney transplantation without disease recurrence”. American Jjournal of Transplantation: Official Journal of the American Society of Transplantation and the American Society of Transplant Surgeons, 2(5), 425–428.
156 Jordan, S. C. et al. (2004). “Evaluation of intravenous immunoglobulin as an agent to lower allosensitization and improve transplantation in highly sensitized adult patients with end-stage renal disease: report of the NIH IG02 trial”. Journal of the American Society of Nephrology: JASN, 15(12), 3256–3262.
157 Tejeda-Strop, A. et al. (2017). “Evaluation of Potencies of Immune Globulin Products against Hepatitis A”. JAMA internal medicine, 177(3), 430-432.
158 Schroeder, H.W. Jr., & Dougherty, C.J. (2012). “Review of intravenous immunoglobulin replacement therapy trials for primary humoral immunodeficiency patients”. Infection, 40(6), 601-611.
159 Ammann, E. M. et al. (2016). “Intravenous immune globulin and thromboembolic adverse events in patients with hematologic malignancy.” Blood, 127(2), 200–207.
160 Tullis, J. L. & Pennell, R. B. (1968). “Transfusion of specific plasma components”. Annual Review of Medicine, 19, 233–246.
161 Foster, P.R. & Bienek, C. (2008) “Fractionated products”. In J.A.J. Barbara, F.A.M. Regan & M.C. Contreras (Eds.), Transfusion Microbiology. Cambridge University Press, 259-303.
162 Watt, J.G. et al. (1972). “New Developments in Large-scale Plasma Fractionation”. Proceedings of the Royal Society of Edinburgh, Section B: Biological Sciences. 71(3), 15-34.
163 Foster, P. R. et al. (1982). “Control of large-scale plasma thawing for recovery of cryoprecipitate Factor VIII”. Vox Sanguinis, 42(4), 180–189.
164 Burnouf T. (1995). “Chromatography in plasma fractionation: benefits and future trends”. Journal of Chromatography. B, Biomedical Applications, 664(1), 3–15.
165 Burnouf T. (2007). “Modern plasma fractionation”. Transfusion Medicine Reviews, 21(2), 101–117.
166 Burnouf T. (2007). “Fractionnement plasmatique international: état des lieux [Plasma fractionation in the world: current status]”. Transfusion Clinique et Biologique: Journal de la Société française de Transfusion Sanguine, 14(1), 41–50.
167 Burnouf, T. (2018). “An overview of plasma fractionation”, Annals of Blood, Vol 3, (33), pp. 1-10.
168 Prevot, J., & Jolles, S. (2020). “Global immunoglobulin supply: steaming towards the iceberg?”. Current Opinion in Allergy and Cinical Immunology, 20(6), 557-564.
169 Johnston, A. & Adcock, W. (2000). “The use of chromatography to manufacture purer and safer plasma products”. Biotechnology & Genetic Engineering Reviews, 17, 37–70. [online] Available at: https:doi.org/10.1080/02648725.2000.10647987.
170 Steinbuch, M. (1982). “New trends in plasma fractionation”. Joint meetings of the 19th Congress of the International Society of Haematology and the 17th Congress of the International Society of Blood Transfusion. Budapest, 214-228.
171 Curling, J.M. (1982, August 1-7). Current practice and future possibilities in plasma protein fractionation. Joint meetings of the 19th Congress of the International Society of Haematology and the 17th Congress of the International Society of Blood Transfusion:Hungary, Budapest.
172 Burnouf, T. (2016). “Current status and new developments in the production of plasma derivatives”. ISBT Science Series, 11(2), 18-25.
173 Burnouf T. (2007). “Modern plasma fractionation”. Transfusion Medicine Reviews, 21(2), 101–117.
174 Johnston, A. & Adcock, W. (2000). “The use of chromatography to manufacture purer and safer plasma products”. Biotechnology & Genetic Engineering Reviews, 17, 37–70. [online] Available at: https:doi.org/10.1080/02648725.2000.10647987.
175 Steinbuch, M. (1982). “New trends in plasma fractionation”. Joint meetings of the 19th Congress of the International Society of Haematology and the 17th Congress of the International Society of Blood Transfusion. Budapest, 214-228.
176 Matejtschuk, P., Dash, C.H. & Gascoigne, E.W. (2000). “Production of human albumin solution: a continually developing colloid”. British Journal of Anaesthesia, 85(6), 887-95.
177 Di Minno, G. et al. (2016). Current concepts in the prevention of pathogen transmission via blood/plasma-derived products for bleeding disorders. Blood Reviews, 30(1), 35–48.
178 Hollinger, F. B. (1990). Hepatitis B Virus. In: B. N. Fields & D. M. Knipe (Eds.), Fields Virology. New York: Raven Press, 2171-2236.
179 Suomela, H. (1993). “Inactivation of viruses in blood and plasma products”. Transfusion medicine reviews, 7(1), 42–57.
180 Recommendations on choice of therapeutic products for the treatment of patients with haemophilia A, haemophilia B and von Willebrand’s disease. (1992). Blood Coagulation & Fibrinolysis, 3(2), 205–214.
181 Hoyer, L.W. (1997). Inhibitors in haemophilia. In: C. D. Forbes, L. M. Aledort & R. Madhok (Eds.), Hemophilia. London, New York: Chapman & Hall, 213-227.
182 Bolton-Maggs, P. H. (2006). “Optimal haemophilia care versus the reality”. British Journal of Haematology, 132(6), 671–682.
183 Schulman, S. et al. (1992). “Transmission of hepatitis C with pasteurised Factor VIII”. Lancet (London, England), 340(8814), 305–306.
184 Pollmann, H. & Jürgens, H. (1992). “Transmission of hepatitis in haemophiliacs”. Lancet (London, England), 340(8822), 793.
185 Mannucci, P. M. (1992). “Outbreak of hepatitis A among Italian patients with haemophilia”. Lancet (London, England), 339(8796), 819.
186 Robinson, S. M., Schwinn, H., & Smith, A. (1992). “Clotting factors and hepatitis A”. Lancet (London, England), 340(8833), 1465.
187 European Agency for the Evaluation of Medicinal Products. (28 March 2001). EMEA Workshop on Viral Safety of Plasma-Derived Medicinal Products with Particular Focus on Non-Enveloped Viruses. EMEA/CPMP/BWP/BPWG/4080/00, London. [online] Available at: https://www.ema.europa.eu/en/documents/report/european-medicines-agency-workshop-viral-safety-plasma-derived-medicinal-products-particular-focus_en.pdf.
188 Mannucci, P. M. (2003). “Hemophilia: treatment options in the twenty-first century”. Journal of Thrombosis and Haemostasis: JTH, 1(7), 1349–1355.
189 Centers for Disease Control and Prevention. (26 November 2019). Pregnancy and Fifth Disease. Centers for Disease Control and Prevention. (November 26, 2019). [online] Available at: https://www.cdc.gov/parvovirusb19/pregnancy.html.
190 Centres for Disease Control and Prevention. (2019). Parvovirus B19 and Fifth Disease. [online] Available at: https://www.cdc.gov/parvovirusb19/fifth-disease.html.
191 Slade, H. B. (1994). “Human Immunoglobulins for intravenous use and hepatitis C viral transmission”. Clinical and Diagnostic Laboratory Immunology, 1(6), 613–619.
192 Tabor, E. (1999). “The epidemiology of virus transmission by plasma derivatives: clinical studies verifying the lack of hepatitis B and C viruses and HIV type 1”. Transfusion, 5, 1160-1168.
193 Yap, P. L. (1996). “The viral safety of intravenous immune globulin”. Clinical and experimental immunology, 104 Suppl 1, 35–42.
194 Centers for Disease Control and Prevention (CDC). (1994). “Outbreak of hepatitis C associated with intravenous immunoglobulin administration--United States, October 1993-June 1994”. MMWR. Morbidity and mortality weekly report, 43(28), 505–509.
195 Farrugia, A. & Quinti, I. (2014). “Manufacture of immunoglobulin products for patients with primary antibody deficiencies – the effect of processing conditions on product safety and efficacy”. Frontiers in immunology, 5, 665.
196 Dixon ,M.S., Acres, S.E. & Ahronheim, G. (1988). “Safety of immune globulin preparations” CMAJ, 139, 522.
197 Piazza, M. et al. (1999). “Prophylaxis of hepatitis C with intramuscular immunoglobulin. Clinical and Economic appraisal.” Biodrugs 12, 292-300.
198 Yei, S., Yu, M.W. & Tankersley, D.L. (1992). “Partitioning hepatitis C virus during Cohn-Oncley fractionation of plasma”. Transfusion, 32, 824-828.
199 Slade, H. B. (1994). “Human Immunoglobulins for intravenous use and hepatitis C viral transmission”. Clinical and diagnostic laboratory immunology, 1(6), 613–619.
200 Tabor, E. (1999). “The epidemiology of virus transmission by plasma derivatives: clinical studies verifying the lack of hepatitis B and C viruses and HIV type 1”. Transfusion, 5, 1160-1168.
201 Yap, P. L. (1996). “The viral safety of intravenous immune globulin”. Clinical and experimental immunology, 104 Suppl 1, 35–42.
202 Bresee, J. S. et al. (1996). “Hepatitis C virus infection associated with administration of intravenous immune globulin. A cohort study”. Journal of American Medical Association, 276(19), 1563–1567.
203 Slade, H. B. (1994). “Human Immunoglobulins for intravenous use and hepatitis C viral transmission”. Clinical and diagnostic laboratory immunology, 1(6), 613–619.
204 Kudesia, G., Price, C., & Clewley, J. (1992). “Passively transfused hepatitis C antibody”. Lancet (London, England), 340(8830), 1291.
205 Council of Europe. (2022). European Pharmacopoeia 10.7. monograph (00557), “Human anti-D immunoglobulin”.
206 Committee for Medicinal Products for Human Use (CHMP). (20 September 2007). “Guideline on the Clinical Investigation of Human Anti-D Immunoglobulin for Intravenous and/or Intramuscular Use”. CPMP/BPWG/575/99 Rev. 1, London. [online] Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-human-anti-d-immunoglobulin-intravenous/intramuscular-use_en.pdf.
207 Fraser, I.D. (1990). “Human Normal anti-D Immunoglobulin – BPL (Intramuscular). Expert report on the clinical documentation”. The National Blood Transfusion Service.
208 SOP No: 302.001.01.NAT. (3 February 1997). The Selection and accreditation of red cell donors/donations for anti-D immunization/boosting.
209 Burckhardt, J. et al. (1988). “Hepatitis-Non-A-Non-B-assoziierte DNA--Nachweis der DNA in gesichert infektiösem anti-D-Immunglobulin [Hepatitis non-A, non-B-associated DNA--demonstration of DNA in proven infectious anti-D-immunoglobulin]”. Immunitat und Infektion, 16(3), 97–99.
210 Dittmann, S. et al. (1991). “Long-term persistence of hepatitis C virus antibodies in a single source outbreak”. Journal of hepatology, 13(3), 323–327.
211 Finlay, T.A. (1997). “Report of the Tribunal of Inquiry into the Blood Transfusion Service Board”. Government of Ireland.
212 Yap, P. L. (1996). “The viral safety of intravenous immune globulin”. Clinical and experimental immunology, 104 Suppl 1, 35–42.
213 Yap, P. L. (1996). “The viral safety of intravenous immune globulin”. Clinical and experimental immunology, 104 Suppl 1, 35–42.
214 Yap, P. L. (1996). “The viral safety of intravenous immune globulin”. Clinical and experimental immunology, 104 Suppl 1, 35–42.
215 Yap, P. L. (1996). “The viral safety of intravenous immune globulin”. Clinical and experimental immunology, 104 Suppl 1, 35–42.
216 Yap, P. L. (1996). “The viral safety of intravenous immune globulin”. Clinical and experimental immunology, 104 Suppl 1, 35–42.
217 Dobson, F. (12 November 1998). New Variant CJD. Hansard, 319, 493-501. [online] Available at: .
218 Seed, C.R. et al. (2018). “Creuttzfeldt-Jakob disease and blood transfusion safety”, Vox Sanguinis, 113, 220-231.
219 Ritchie, D.L., Peden, A. & Barria, M.A. (2021). “Variant CJD: Reflections a Quarter of a Century on”. Pathogens, 10(11), 1413. [online] Available at: https://mdpi.com/journal/pathogens.
220 European Centre for Disease Prevention and Control (ECDC). (3 August 2021). The risk of variant Creutzfeldt-Jakob disease transmission via blood and plasma-derived medicinal products manufactured from donations obtained in the United-Kingdom. ECDC: Stockholm.
221 Medicines & Healthcare products Regulatory Agency (MHRA). (2021). Use of UK plasma for the manufacture of immunoglobulins and vCJD risk.
222 Medicines & Healthcare products Regulatory Agency (MHRA). (2021). Use of UK plasma for the manufacture of immunoglobulins and vCJD risk.
223 European Centre for Disease Prevention and Control (ECDC). (3 August 2021). The risk of variant Creutzfeldt-Jakob disease transmission via blood and plasma-derived medicinal products manufactured from donations obtained in the United-Kingdom.ECDC: Stockholm.
224 Cohn, E.J. et al. (1946). “Preparation and Properties of Serum and Plasma Proteins. IV. A System for the Separation into Fractions of the Protein and Lipoprotein Components of Biological Tissues and Fluids”. Journal of the American Chemical Society, 68(3), 459-475.
225 Minot, G. R., & Taylor, F. H. (1947). “Hemophilia; the clinical use of antihemophilic globulin”. Annals of internal medicine, 26(3), 363–367.
226 Kasper, C. K. (2012). “Judith Graham Pool and the discovery of cryoprecipitate”. Haemophilia: the Official Journal of the World Federation of Hemophilia, 18(6), 833–835.
227 Sibinga, C. T. (1996). “Emile Rémigy and the discovery of anti-haemophilic activity in cryoprecipitate”. Haemophilia: the Official Journal of the World Federation of Hemophilia, 2(1), 56–60.
228 Berntorp, E. (1997). Factor VIII Concentrates. In: C. D. Forbes, L. Aledort & R. Madhok (Eds.), Hemophilia. London, New York: Chapman & Hall, 181-192.
229 McMillan, C. W., Diamond, L. K., & Surgernor R. D. M. (1961). “Treatment of classic hemophilia: the use of fibrinogen rich in Factor VIII for hemorrhage and for surgery”. The New England Journal of Medicine, 265.
230 Pool, J.G., Hershgold, E.J. & Pappenhagen, A.R. (1964). “High-potency antihaemophilic factor concentrate prepared from cryoglobulin precipitate”. Nature, 203(4942), 312.
231 Pool, J.G. & Shannon, A.E. (1965). “Production of high-potency concentrates of antihemophilic globulin in a closed-bag system”. The New England Journal of Medicine, 273(27), 1443-1447.
232 Newman, J. et al. (1971). “Methods for the production of clinically effective intermediate- and high-purity Factor-VIII concentrates”. British Journal of Haematology, 21(1), 1–20.
233 Callum, J.L., Keyvan, K. & Lin, Y. (2009). “Cryoprecipitate: The Current State of Knowledge”. Transfusion Medicine Reviews, 23(3), 177-188.
234 Trinchero, A., Sholzberg, M. & Matino, D. (2020). “The Evolution of Hemophilia Care: Clinical and Laboratory Advances, Opportunities, and Challenges”. Hamostaseologie, 40(3), 311–321.
235 Darby, S.C. et al. (2007). Mortality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood, [online] 110(3), pp.815–825. Available at: https://pubmed.ncbi.nlm.nih.gov/17446349/.
236 Johnson, A. J., Mathews, R. W., & Fulton, A. J. (1984). “Approaches to plasma fractionation for improved recovery and the development of potentially useful clinical factors”. Clinics in Haematology, 13(1), 3–15.
237 Kasper, C. K. (2012). “Judith Graham Pool and the discovery of cryoprecipitate”. Haemophilia: the Official Journal of the World Federation of Hemophilia, 18(6), 833–835.
238 Bidwell, E. (1955). “The purification of antihaemophilic globulin from animal plasma”. British Journal of Haematology, 1(4), 386–389.
239 Biggs R. & MacFarlane R.G. (Eds.) (1966). Treatment of Haemophilia and Other Coagulation Diseases. Oxford: Blackwell.
240 Morfini, M. et al.(2013). “Clinical use of Factor VIII and Factor IX concentrates”. Blood Transfusion, 11 Suppl 4 (Suppl 4), s55–s63.
241 Didisheim, P. et al.(1959). “Preparation of a human plasma fraction rich in prothrombin, proconvertin, Stuart Factor, and PTC and a study of its activity and toxicity in rabbits and man”. The Journal of Laboratory and Clinical Medicine, 53(2), 322–330.
242 Kekwick, R. A., & Mackay, M. E. (1954). “The separation of protein fractions from human plasma with ether”. Special Report Series [Medical Research Council (Great Britain)], 286, 1–75.
243 Franchini M. (2013). “The modern treatment of haemophilia: a narrative review”. Blood Transfusion, 11(2), 178–182.
244 Rizza C. R. (1995). “The first patient to receive Factor IX concentrate in the UK: a recollection”. Haemophilia: the Official Journal of the World Federation of Hemophilia, 1(3), 210–212.
245 Foster, P. R. et al. (1982). “Control of large-scale plasma thawing for recovery of cryoprecipitate Factor VIII”. Vox Sanguinis, 42(4), 180–189.
246 Watt, J.G. et al. (1972). “New Developments in Large-scale Plasma Fractionation”. Proceedings of the Royal Society of Edinburgh, Section B: Biological Sciences. 71(3), 15-34.
247 Curling, J.M. (1982, August 1-7). Current practice and future possibilities in plasma protein fractionation. Joint meetings of the 19th Congress of the International Society of Haematology and the 17th Congress of the International Society of Blood Transfusion: Hungary, Budapest.
248 Schneider, W. et al. (1975). “An alternative method of large scale plasma fractionation for the isolation of serum albumin”. Blut Zeitschrift für die Gesamte Blutforschung , 30(2), 121-134.
249 Friedli, H. et al. (1976). “Studies on New Process Procedures in Plasma Fractionation on an Industrial Scale”. Vox Sanguinis, 31(4), 289-295.
250 Tullis, J. L. & Pennell, R. B. (1968). “Transfusion of specific plasma components”. Annual Review of Medicine, 19, 233–246.
251 Foster, P.R. & Bienek, C. (2008) “Fractionated products”. In J.A.J. Barbara, F.A.M. Regan & M.C. Contreras (Eds.), Transfusion Microbiology. Cambridge University Press, 259-303.
252 Foster, P. R. et al. (1982). “Control of large-scale plasma thawing for recovery of cryoprecipitate Factor VIII”. Vox Sanguinis, 42(4), 180–189.
253 Burnouf T. (2007). “Modern plasma fractionation”. Transfusion Medicine Reviews, 21(2), 101–117.
254 Gunson, H.H. (1979). Variables Involved in Cryoprecipitate Production and their Effect on Factor VIII Activity. British Journal of Haematology, 43(2), 287–295.
255 Kasper, C. K. et al. (1975). “Determinants of Factor VIII recovery in cryoprecipitate”. Transfusion, 15(4), 312–322.
256 Mason, E. C. (1978). “Thaw-siphon technique for production of cryoprecipitate concentrate of Factor VIII”. Lancet (London, England), 2(8079), 15–17.
257 Rock, G. A. et al. (1979). “Improved yields of Factor VIII from heparinized plasma”. Vox Sanguinis, 36(5), 294–300.
258 McLeod, B. C. et al. (1988). “Long-term frequent plasma exchange donation of cryoprecipitate”. Transfusion, 28(4), 307–310.
259 Berntorp, E. (1997). Factor VIII Concentrates. In: C. D. Forbes, L. Aledort & R. Madhok (Eds.), Hemophilia. London, New York: Chapman & Hall, 181-192.
260 Butenas, S. et al. (2009). “Potency and mass of Factor VIII in FVIII products”. Haemophilia, 15(1), 63–72.
261 Marlar, R.A. et al. (2020). Clinical utility and impact of the use of the chromogenic vs one-stage factor activity assays in haemophilia A and B. European Journal of Haematology, 104(1), pp.3–14. [online] Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6916414/.
262 Barrowcliffe, T. W. et al. (2002). “Coagulation and chromogenic assays of Factor VIII activity: general aspects, standardization, and recommendations”. Seminars in Thrombosis and Hemostasis, 28(3), 247–256.
263 Butenas, S. et al. (2009). “Potency and mass of Factor VIII in FVIII products”. Haemophilia, 15(1), 63–72.
264 Berntorp, E. (1997). Factor VIII Concentrates. In: C. D. Forbes, L. Aledort & R. Madhok (Eds.), Hemophilia. London, New York: Chapman & Hall, 181-192.
265 Morfini M. (2017). “The History of Clotting Factor Concentrates Pharmacokinetics”. Journal of Clinical Medicine, 6(3), 35.
266 Kreil, T.R. (2018). “Building blocks of the viral safety margins of industrial plasma products”. Annals of Blood, 3(14). [online] Available at: http://dx.doi.org/10.21037/aob.2018.02.01.
267 World Health Organization. (2019). World Health Organization Model List of Essential Medicines: 21st List. [online] Available at: https://apps.who.int/iris/handle/10665/325771.
268 Schramm, W. et al. (2012). “Haemophilia care in Europe: the ESCHQoL study”. Haemophilia, 18(5), 729–737.
269 Pollak, E.S. & Nagalla, S. (Updated March 2021). “von Willebrand Disease Treatment & Management”. Medscape. [online] Available at: https://emedicine.medscape.com/article/206996-treatment.
270 Cochrane Injuries Group Albumin Reviewers. (1998). “Human albumin administration in critically ill patients: systematic review of randomised controlled trials”. BMJ (Clinical research ed.), 317(7153), 235–240.
271 Haynes, G. R., Navickis, R. J., & Wilkes, M. M. (2003). “Albumin administration – what is the evidence of clinical benefit? A systematic review of randomized controlled trials”. European Journal of Anaesthesiology, 20(10), 771–793.
272 Finfer, S. et al. (2003). “Efficacy of albumin in critically ill patients”. BMJ (Clinical research ed.), 326(7389), 559–560.
273 Finfer, S. et al. (2004). “A comparison of albumin and saline for fluid resuscitation in the intensive care unit”. The New England Journal of Medicine, 350(22), 2247–2256.
274 Myburgh, J. A., & Mythen, M. G. (2013). “Resuscitation fluids”. The New England Journal of Medicine, 369(13), 1243–1251.
275 Stoller, J. K., & Aboussouan, L. S. (2012). “A review of α1-antitrypsin deficiency”. American Journal of Respiratory and Critical Care Medicine, 185(3), 246–259.
276 Rodgers, G. M. (2009). “Role of antithrombin concentrate in treatment of hereditary antithrombin deficiency. An update”. Thrombosis and Haemostasis, 101(5), 806–812.
277 Hofstra, J. J. et al. (2012). “Treatment of hereditary angioedema with nanofiltered C1-esterase inhibitor concentrate (Cetor®): multi-center phase II and III studies to assess pharmacokinetics, clinical efficacy and safety”. Clinical Immunology (Orlando, Fla.), 142(3), 280–290.
278 Bruton, O.C. (1952). “Agammaglobulinemia”. Pediatrics, 9(6), 722-728.
279 Barandun, S., & Isliker, H. (1986). “Development of immunoglobulin preparations for intravenous use”. Vox Sanguinis, 51(2), 157–160.
280 Imbach, P. et al. (1981). “High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood”. Lancet, 1(8232), 1228–1231.
281 Studies on evidence-based therapeutic use of intravenous immunoglobulin in auto-immune and inflammatory diseases. [online] Available at: http://www.cochrane.org/.
282 Sewell, W. A. et al. (2014). “European consensus proposal for immunoglobulin therapies”. European Journal of Immunology, 44(8), 2207–2214.
283 Provan, D. et al. (2008). “Prescribing intravenous immunoglobulin: summary of Department of Health guidelines”. BMJ (Clinical research ed.), 337, a1831.
284 Smith, S. D., Dennington, P. M., & Cooper, A. (2010). “The use of intravenous immunoglobulin for treatment of dermatological conditions in Australia: A review”. The Australasian Journal of Dermatology, 51(4), 227–237.
285 Demeyere, R. et al. (2010). “Comparison of fresh frozen plasma and prothrombin complex concentrate for the reversal of oral anticoagulants in patients undergoing cardiopulmonary bypass surgery: a randomized study”. Vox Sanguinis, 99(3), 251–260.
286 Lechner, K. et al. (1979). “Effect of treatment with activated prothrombin complex concentrate (FEIBA) on Factor VIII-antibody level”. Thrombosis and Haemostasis, 40(3), 478–485.
287 Peyvandi, F. et al. (2016). “A Randomized Trial of Factor VIII and Neutralizing Antibodies in Hemophilia A”. The New England Journal of Medicine, 374(21), 2054–2064.
288 Godschalk, P. C. et al. (2004). “The crucial role of Campylobacter jejuni genes in anti-ganglioside antibody induction in Guillain-Barre syndrome”. The Journal of Clinical Investigation, 114(11), 1659–1665.
289 Turgeon, A. F. et al. (2007). “Meta-analysis: intravenous immunoglobulin in critically ill adult patients with sepsis”. Annals of Internal Medicine, 146(3), 193–203.
290 Anthony, R. M. et al. (2011). “Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway”. Nature, 475(7354), 110–113.
291 Kaneko, Y., Nimmerjahn, F., & Ravetch, J. V. (2006). “Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation”. Science (New York, N.Y.), 313(5787), 670–673.
292 Over, J. et al. (1980). “Heterogeneity of human Factor VIII. III. Transitions between forms of Factor VIII present in cryoprecipitate and in cryosupernatant plasma”. The Journal of Laboratory and Clinical Medicine, 95(3), 323–334.
293 Di Giambattista, M. et al. (2007). “In silico prediction of FVIII epitopes recognised by natural autoantibodies in polyvalent immunoglobulin concentrates”. Molecular Immunology, 44(8), 1903–1913.
294 Grancha, S. et al. (2013), “Factor IX”. In J. Bertolini, N. Goss & J. Curling (Eds.), Production of Plasma Proteins for Therapeutic Use, First Edition, 81-92.
295 Di Giambattista. (2007). “In Silico Prediction of FVIII Epitopes Recognised by Natural Autoantibodies in Intermediate Fractions and Intravenous Immunoglobulins from Large Plasma Pools”. Plasma Product Biotechnology Meeting 2007, Fifth International Meeting, 8-12 May 2007, Hermitage/Biodola, Elba, Italy.
296 Scott, D. W., & Pratt, K. P. (2020). “Factor VIII: Perspectives on Immunogenicity and Tolerogenic Strategies”. Frontiers in Immunology, 10, 3078. [online] Available at: www.frontier.org.
297 Lacroix-Desmazes, S. et al. (2020). “Tolerating Factor VIII: Recent Progress”. Frontiers in Immunology, 10, 2991. [online] Available at: www.frontier.org.
298 Astermark, J. (2015). “FVIII inhibitors: pathogenesis and avoidance”. Blood, 125, 2045-2051.
299 Eeckardt, C.L. et al. (2011). “Surgery and inhibitor development in hemophilia A: a systemic review”. Journal of Thrombosis and Haemostasis, 9(10), 1948–1958.
300 Witmer, C., & Young, G. (2013). “Factor VIII inhibitors in hemophilia A: rationale and latest evidence”. Therapeutic Advances in Hematology, 4(1), 59–72.
301 The European Agency for the Evaluation of Medicinal Products, Committee for Proprietary Medicinal Products. (1996). “Note for guidance on virus validation studies: the design, contribution and interpretation of studies validating the inactivation and removal of viruses”, CPMP/BWP/268/95. [online] Available at: http://wwweurope.eu/docs/en_GB/document_library/Scientific_guideline wc 50000 3684.pdf2009/09/.
302 EMEA/CPMP/ 269/95 rev.3 ( 25 January 2001), “Note for Guidance on Plasma-Derived Medicinal Products”. [online] Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/note-guidance-plasma-derived-medicinal-products_en.pdf.
303 Peerlinck, K. et al. (1997). “Factor VIII inhibitors in previously treated haemophilia A patients with a double virus-inactivated plasma derived Factor VIII concentrate”. Thrombosis and Haemostasis, 77(1), 80–86.
304 Rosendaal, F. R. et al. (1993). “A sudden increase in Factor VIII inhibitor development in multitransfused hemophilia A patients in The Netherlands. Dutch Hemophilia Study Group”. Blood, 81(8), 2180–2186.
305 Barrowcliffe, T. W., Kemball-Cook, G., & Tubbs, J. E. (1993). “Inhibitor development and activated Factor VIII in concentrates”. Thrombosis and Haemostasis, 70(6), 1065–1066.
306 Sawamoto, Y. et al. (1998). “Dominant C2 domain epitope specificity of inhibitor antibodies elicited by a heat pasteurized product, Factor VIII CPS-P, in previously treated hemophilia A patients without inhibitors”. Thrombosis and Haemostasis, 79(1), 62–68.
307 Peerlinck, K. et al. (1997). “Factor VIII inhibitors in previously treated haemophilia A patients with a double virus-inactivated plasma derived Factor VIII concentrate”. Thrombosis and Haemostasis, 77(1), 80–86.
308 Laub, R. et al. (1999). “Inhibitors in German hemophilia A patients treated with a double virus inactivated Factor VIII concentrate bind to the C2 domain of FVIII light chain”. Thrombosis and Haemostasis, 81(1), 39–44.
309 Raut, S. et al. (1998). “Modification of Factor VIII in therapeutic concentrates after virus inactivation by solvent-detergent and pasteurisation”. Thrombosis and Haemostasis, 80(4), 624–631.
310 Lusher, J. et al. (2004). “Human recombinant DNA-derived antihemophilic factor in the treatment of previously untreated patients with hemophilia A: final report on a hallmark clinical investigation”. Journal of Thrombosis and Haemostasis, 2(4), 574–583.
311 Ehrenforth, S. et al. (1992). Incidence of development of Factor VIII and Factor IX inhibitors in haemophiliacs. The Lancet, 339(8793), 594–598.
312 Addiego, J. et al. (1993). Frequency of inhibitor development in haemophiliacs treated with low-purity Factor VIII. The Lancet, 342(8869), 462–464.
313 Lusher, J.M. (2000). Inhibitors in young boys with haemophilia. Best Practice & Research Clinical Haematology, 13(3), 457–468.
314 DiMichele, D. et al. (1999). Utilization of Previously Treated Patients (PTPs), Noninfected Patients (NIPs), and Previously Untreated Patients (PUPs) in the Evaluation of New Factor VIII and Factor IX Concentrates. Thrombosis and Haemostasis, 81(03), 462–462.
315 Hoyer, L.W. (1997). Inhibitors in haemophilia. In: C. D. Forbes, L. M. Aledort & R. Madhok (Eds.), Hemophilia. London, New York: Chapman & Hall, 213-227.
316 Lusher, J. M. (2004). “Is the incidence and prevalence of inhibitors greater with recombinant products? No.” Journal of Thrombosis and Haemostasis, 2(6), 863–865.
317 Darby, S. C. et al. (2004). “The incidence of Factor VIII and Factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99”. Journal of Thrombosis and Haemostasis, 2(7), 1047–1054.
318 Farrugia, A., Manno, C. S., & Evatt, B. L. (2004). “Emerging and receding risks of therapeutic regimens for haemophilia”. Haemophilia : the Official Journal of the World Federation of Hemophilia, 10 Suppl 4, 47–54.
319 Saint-Remy, J. M., Lacroix-Desmazes, S., & Oldenburg, J. (2004). “Inhibitors in haemophilia: pathophysiology”. Haemophilia: the Official Journal of the World Federation of Hemophilia, 10 Suppl 4, 146–151.
320 Peerlinck, K. et al. (1993). “A higher than expected incidence of Factor VIII inhibitors in multitransfused haemophilia A patients treated with an intermediate purity pasteurized Factor VIII concentrate”. Thrombosis and Haemostasis, 69(2), 115–118.
321 Peerlinck, K. et al. (1997). “Factor VIII inhibitors in previously treated haemophilia A patients with a double virus-inactivated plasma derived Factor VIII concentrate”. Thrombosis and Haemostasis, 77(1), 80–86.
322 Rosendaal, F. R. et al. (1993). “A sudden increase in Factor VIII inhibitor development in multitransfused hemophilia A patients in The Netherlands. Dutch Hemophilia Study Group”. Blood, 81(8), 2180–2186.
323 Lusher, J. M. et al. (1993). “Recombinant Factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. Kogenate Previously Untreated Patient Study Group”. The New England Journal of Medicine, 328(7), 453–459.
324 Hoyer, L.W. (1997). Inhibitors in haemophilia. In: C. D. Forbes, L. M. Aledort & R. Madhok (Eds.), Hemophilia. London, New York: Chapman & Hall, 213-227.
325 Hay, C. R. (1991). “Innovative use of porcine Factor VIII:C for immune tolerance induction”. The American Journal of Medicine, 91(5A), 27S–29S.
326 Mizon, P. et al. (1992). “Myocardial infarction after FEIBA therapy in a hemophilia-B patient with a Factor IX inhibitor”. Annals of Hematology, 64(6), 309–311.
327 Levi, M. et al. (2010). “Safety of recombinant activated Factor VII in randomized clinical trials”. The New England Journal of Medicine, 363(19), 1791–1800.
328 Watson, H. G., & Ludlam, C. A. (1992). “Immunological abnormalities in haemophiliacs”. Blood Reviews, 6(1), 26–33.
329 Eichinger, S. et al. (1992). “Factor VIII concentrates in HIV-1-positive hemophiliacs--is pure better?”. Haemostasis, 22(1), 25–31.
330 Giangrande, P. L. (2003). “Adverse events in the prophylaxis of haemophilia”. Haemophilia: the Official Journal of the World Federation of Hemophilia, 9(Suppl 1), 50–56.
331 Suzuki, N. et al. (2003). “Successful induction of immune tolerance in a patient with haemophilia B with inhibitor”. Haemophilia: the Official Journal of the World Federation of Hemophilia, 9(3), 340–342.
332 Knight, A.I., Haines, J., & Zuber, S., (2013). “Thermal inactivation of animal virus pathogens”, Current Topics in Virology, 11, 103-119.
333 Mani, B. et al. (2007). “Molecular mechanism underlying B19 virus inactivation and comparison to other parvoviruses”. Transfusion, 47(10), 1765–1774.
334 Boschetti, N. et al. (2005). “Virus safety of intravenous immunoglobulin: future challenges”. Clinical Reviews in Allergy and Immunology, 29(3), 333–344.
335 Franks, F. (1988) Conformational Stability Denaturation and Renaturation. In: F. Franks (Eds.), Characterization of Proteins. Humana Press, 95-126.
336 Eibl, J. et al. (1988). “Ways to reduce the risk of transmission of viral infections by plasma and plasma products” Vox Sanguinis, 54, 228-245.
337 The European Agency for the Evaluation of Medicinal Products, Committee for Proprietary Medicinal Products. (1996). “Note for guidance on virus validation studies: the design, contribution and interpretation of studies validating the inactivation and removal of viruses”, CPMP/BWP/268/95. [online] Available at: http://wwweurope.eu/docs/en_GB/document_library/Scientific_guideline wc 50000 3684.pdf2009/09/.
338 EMEA/CPMP/ 269/95 rev.3 ( 25 January 2001), “Note for Guidance on Plasma-Derived Medicinal Products”. [online] Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/note-guidance-plasma-derived-medicinal-products_en.pdf.
339 Prince, A.M. et al. (1987), “The Development of Virus-Free Labile Blood Derivatives – A Review”, European Journal of Epidemiology, 3(2), 103-118.
340 Mannucci, P. M. (1993). “Clinical evaluation of viral safety of coagulation Factor VIII and IX concentrates”. Vox Sanguinis, 64(4), 197–203.
341 Cai, K. et al. (2005). “Ensuring the biologic safety of plasma-derived therapeutic proteins: detection, inactivation, and removal of pathogens”. BioDrugs: clinical immunotherapeutics, biopharmaceuticals and gene therapy, 19(2), 79–96.
342 Berntorp, E. et al. (2008). “A systematic overview of the first pasteurised VWF/FVIII medicinal product, Haemate P/ Humate -P: history and clinical performance”. European Journal of Haematology. Supplementum, (70), 3–35.
343 Klamroth, R., Gröner, A., & Simon, T. L. (2014). “Pathogen inactivation and removal methods for plasma-derived clotting factor concentrates”. Transfusion, 54(5), 1406–1417.
344 Buchacher, A. &Iberer, G. (2006). “Purification of intravenous immunoglobulin G from human plasma-aspects of yield and virus safety”. Biotechnology Journal, 1(2), 148-163.
345 European Agency for the Evaluation of Medicinal Products. (28 March 2001). EMEA Workshop on Viral Safety of Plasma-Derived Medicinal Products with Particular Focus on Non-Enveloped Viruses. EMEA/CPMP/BWP/BPWG/4080/00, London. [online] Available at: https://www.ema.europa.eu/en/documents/report/european-medicines-agency-workshop-viral-safety-plasma-derived-medicinal-products-particular-focus_en.pdf.
346 Dähnert, L. et al. (2021). “Hepatitis E virus: Efficacy of pasteurization of plasma-derived VWF/FVIII concentrate determined by pig bioassay”. Transfusion, 61(4), 1266–1277.
347 Heimburger, N., Schwinn, H. & Mauler, R., (1980) “Factor VIII concentrate – now free from Hepatitis risk: progress in the treatment of haemophilia”. Die gelben Hefte, 4, 165-174. [online] Available at: https://www.penroseinquiry.org.uk/finalreport/pdf/SNB0045880.PDF.
348 Heimburger, N. et al. (1981). “A Factor VIII Concentrate, Highly Purified and Heated in Solution”. Pathophysiology of Haemostasis and Thrombosis, 10, 204.
349 Schimpf, K. et al. (1987). “Absence of Hepatitis after treatment with a pasteurized Factor VIII concentrate in patients with hemophilia and no previous transfusions”. New England Journal of Medicine, 316, 918-929.
350 Schimpf, K. et al. (1989). Absence of Anti-Human Immunodeficiency Virus Types 1 and 2 Seroconversion after the Treatment of Hemophilia A or von Willebrand’s Disease with Pasteurized Factor VIII Concentrate. New England Journal of Medicine, 321(17), 1148–1152.
351 Berntorp, E. et al. (2008). “A systematic overview of the first pasteurized VWF/FVIII medicinal product, Haemate P/Humate-P: history and clinical performance”. European Journal of Haematology, 80(Suppl 70), 3-35.
352 Gröner, A. et al. (2018). “Effective inactivation of a wide range of viruses by pasteurization”. Transfusion, 58(1), 41–51.
353 Berntorp, E. et al. (2008). “A systematic overview of the first pasteurised VWF/FVIII medicinal product, Haemate P/ Humate -P: history and clinical performance”. European journal of Haematology. Supplementum, (70), 3–35.
354 Ng, P. K., & Dobkin, M. B. (1985). “Pasteurization of antihemophilic factor and model virus inactivation studies”. Thrombosis research, 39(4), 439–447.
355 Ofosu, F. A., Freedman, J., & Semple, J. W. (2008). “Plasma-derived biological medicines used to promote haemostasis”. Thrombosis and Haemostasis, 99(5), 851–862.
356 Gröner, A. et al. (2018). “Effective inactivation of a wide range of viruses by pasteurization”. Transfusion, 58(1), 41–51.
357 Menache, D., (1985). “Measures to Inactivate Viral Contaminants of Pooled Plasma Products”, Infection, Immunity and Blood Transfusion, 407-423.
358 Remington, K. M. et al. (2004). “Inactivation of West Nile virus, vaccinia virus and viral surrogates for relevant and emergent viral pathogens in plasma-derived products”. Vox Sanguinis, 87(1), 10–18.
359 “EMEA Workshop on Viral Safety of Plasma-Derived Medicinal Products with Particular Focus on Non-Enveloped Viruses”. European Agency for the Evaluation of Medicinal Products (28 March 2001). EMEA/CPMP/BWP/BPWG/4080/00, London. Available online: .
360 Vermeer, A. W., & Norde, W. (2000). “The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein”. Biophysical Journal, 78(1), 394–404.
361 Kempf, C., Stucki, M., & Boschetti, N. (2007). “Pathogen inactivation and removal procedures used in the production of intravenous immunoglobulins”. Biologicals: Journal of the International Association of Biological Standardization, 35(1), 35–42.
362 Eibl, J. et al. (1988). “Ways to reduce the risk of transmission of viral infections by plasma and plasma products. A comparison of methods, their advantages and disadvantages”. Vox Sanguinis, 54(4), 228–245.
363 Cai, K. et al. (2005). “Ensuring the biologic safety of plasma-derived therapeutic proteins: detection, inactivation, and removal of pathogens”. BioDrugs: Clinical Immunotherapeutics, Biopharmaceuticals and Gene Therapy, 19(2), 79–96.
364 Mannucci, P. M. et al. (1992). “Low risk of viral infection after administration of vapor-heated Factor VIII concentrate. International Investigator Group”. Transfusion, 32(2), 134–138.
365 Barrett, P. N. et al. (1997). “Inactivation of hepatitis A virus in plasma products by vapor heating”. Transfusion, 37(2), 215–220.
366 Eibl, J. et al. (1988). “Ways to reduce the risk of transmission of viral infections by plasma and plasma products. A comparison of methods, their advantages and disadvantages.” Vox Sanguinis, 54, 228-245.
367 Prince, A.M. et al. (1987), “The Development of Virus-Free Labile Blood Derivatives – A Review”, European Journal of Epidemiology, 3(2), 103-118.
368 Prince, A., & Brotman, B. (2001). “Perspectives on hepatitis B studies with chimpanzees”. ILAR Journal, 42(2), 85-88.
369 Prince, A.M. et al. (1987), “The Development of Virus-Free Labile Blood Derivatives – A Review”, European Journal of Epidemiology, 3(2), 103-118.
370 Farrugia, A. (2017). “Guide for the assessment of clotting factor concentrates”, World Federation of Hemophilia, (3rd edition). [online] Available at: https://www1.wfh.org/publication/files/pdf-1271.pdf.
371 Kasper, C. K., & Lusher, J. M. (1993). “Recent evolution of clotting factor concentrates for hemophilia A and B. Transfusion Practices Committee”. Transfusion, 33(5), 422–434.
372 Foster, P.R. (1997). Safer Clotting Factor Concentrates. Hemophilia. London, New York: Chapman & Hall, 307-332.
373 Winkelman, L. et al. (1989). “Severely heated therapeutic Factor VIII concentrate of high specific activity”. Vox Sanguinis, 57(2), 97–103.
374 Roberts, P. L. et al. (2007). “Effect of manufacturing process parameters on virus inactivation by dry heat treatment at 80 degrees C in Factor VIII”. Vox Sanguinis, 92(1), 56–63.
375 The European Medicines Agency, Committee for medicinal products for human use. (2011). “Guideline on Plasma-Derived Medicinal Products”, EMA/CHMP/BWP/706271/2010. [online] Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-plasma-derived-medicinal-products_en.pdf.
376 Klamroth, R., Gröner, A., & Simon, T. L. (2014). “Pathogen inactivation and removal methods for plasma-derived clotting factor concentrates”. Transfusion, 54(5), 1406–1417.
377 “EMEA Workshop on Viral Safety of Plasma-Derived Medicinal Products with Particular Focus on Non-Enveloped Viruses”. European Agency for the Evaluation of Medicinal Products (28 March 2001). EMEA/CPMP/BWP/BPWG/4080/00, London. Available online: .
378 Leveton, L.B., Sox Jr., H.C., Stoto, M.A. (Eds.). (1995). HIV and the Blood Supply – An Analysis of Crisis Decision Making. Committee to Study HIV Transmission Through Blood and Blood Products, Division of Health Promotion and Disease Prevention, Institute of Medicine, National Academy Press, Washington Academy Press.
379 Foster, P.R., (2015) PEN .013.1330. [online] Available at: https://penroseinquiry.org.uk/. [Accessed 23 February 2021].
380 Dichtelmüller, H. et al. (1996). “Improvement of virus safety of a S/D-treated Factor VIII concentrate by additional dry heat treatment at 100 degrees C”. Biologicals: Journal of the International Association of Biological Standardization, 24(2), 125–130.
381 Benny, A. G. et al. (1988). “Influence of heat treatment on FVIII:C recovery from freeze dried cryoprecipitate”. Journal of Clinical Pathology, 41(9), 945–947.
382 Skjønsberg, O. H. et al. (1987). “Characteristics of a heat treated antihaemophilic cryoprecipitate”. Thrombosis Research, 45(5), 625–634.
383 Mazurier, C. et al. (1987). “In vitro and in vivo evaluation of a Factor VIII concentrate heat-treated to inactivate HTLV-III/LAV viruses. Favourable effects of heating on the von Willebrand factor”. Vox Sanguinis, 52(4), 265–271.
384 Messori, A. et al. (1992). “Pharmacokinetics of two pasteurized Factor VIII concentrates by different and multicenter assays of factor VIII activity”. Thrombosis Research, 65(6), 699–708.
385 Stampe, D., Wieland, B., & Köhle, A. (1988). “Preparation and heat-treatment of Factor-IX-concentrates”. Folia Haematol Int Mag Klin Morphol Blutforsch, 115(4), 523–527.
386 Farrugia, A. et al. (1989). “Effects of plasma collection systems and processing parameters on the quality of Factor IX concentrate”. Vox Sanguinis, 57(1), 4–9.
387 The European Medicines Agency, Committee for medicinal products for human use. (2011). “Guideline on Plasma-Derived Medicinal Products”, EMA/CHMP/BWP/706271/2010. [online] Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-plasma-derived-medicinal-products_en.pdf.
388 Butler, S. L., & Falke, J. J. (1996). “Effects of protein stabilizing agents on thermal backbone motions: a disulfide trapping study”. Biochemistry, 35(33), 10595–10600.
389 Smales, C. M., Pepper, D. S., & James, D. C. (2000). “Protein modification during antiviral heat bioprocessing”. Biotechnology and Bioengineering, 67(2), 177–188.
390 Leveton, L.B., Sox Jr., H.C., Stoto, M.A. (Eds.). (1995). HIV and the Blood Supply – An Analysis of Crisis Decision Making. Committee to Study HIV Transmission Through Blood and Blood Products, Division of Health Promotion and Disease Prevention, Institute of Medicine, National Academy Press, Washington Academy Press.
391 Menache, D., (1985). “Measures to Inactivate Viral Contaminants of Pooled Plasma Products”, Infection, Immunity and Blood Transfusion, 407-423.
392 Menache, D., (1985). “Measures to Inactivate Viral Contaminants of Pooled Plasma Products”, Infection, Immunity and Blood Transfusion, 407-423.
393 Prince, A., & Brotman, B. (2001). “Perspectives on hepatitis B studies with chimpanzees”. ILAR Journal, 42(2), 85-88.
394 Pool, J.G. & Shannon, A.E. (1965). “Production of high-potency concentrates of antihemophilic globulin in a closed-bag system”. The New England Journal of Medicine, 273(27), 1443-1447.
395 Butenas, S. et al. (2009). “Potency and mass of Factor VIII in FVIII products”. Haemophilia, 15(1), 63–72.
396 Prince, A. M. et al. (1984). “Inactivation of hepatitis B and Hutchinson strain non-A, non-B hepatitis viruses by exposure to Tween 80 and ether”. Vox Sanguinis, 46(1), 36–43.
397 Horowitz, B. et al. (1985). “Inactivation of viruses in labile blood derivatives. I. Disruption of lipid-enveloped viruses by tri(n-butyl)phosphate detergent combinations”. Transfusion, 25(6), 516–522.
398 Horowitz B. (1990). “Blood protein derivative viral safety: observations and analysis”. The Yale Journal of Biology and Medicine, 63(5), 361–369.
399 Prince, A.M. et al. (1987), “The Development of Virus-Free Labile Blood Derivatives – A Review”, European Journal of Epidemiology, 3(2), 103-118.
400 Prince, A., & Brotman, B. (2001). “Perspectives on hepatitis B studies with chimpanzees”. ILAR Journal, 42(2), 85-88.
401 Prince, A. M. et al. (1984). “Inactivation of hepatitis B and Hutchinson strain non-A, non-B hepatitis viruses by exposure to Tween 80 and ether”. Vox Sanguinis, 46(1), 36–43.
402 Horowitz, B. et al. (1985). “Inactivation of viruses in labile blood derivatives. I. Disruption of lipid-enveloped viruses by tri(n-butyl)phosphate detergent combinations”. Transfusion, 25(6), 516–522.
403 Prince, A.M. et al. (1985). “Quantitative assays for evaluation of HTLV-III: sodium cholate and beta-propiolactone.” Cancer Research, 45 (suppl), 4592s-4594s.
404 Prince, A.M.(1986). “Sterilisation of hepatitis and HTLV-III viruses by exposure to tri(n-butyl)phosphate and sodium cholate”. The Lancet, 327(8483), 706-710.
405 Horowitz B. (1989). “Investigations into the application of tri(n-butyl)phosphate/detergent mixtures to blood derivatives”. Current Studies in Hematology and Blood Transfusion, (56), 83–96.
406 Dichtelmüller, H. O. et al. (2009). “Robustness of solvent/detergent treatment of plasma derivatives: a data collection from Plasma Protein Therapeutics Association member companies”. Transfusion, 49(9), 1931–1943.
407 The European Agency for the Evaluation of Medicinal Products, Committee for Proprietary Medicinal Products. (1996). “Note for guidance on virus validation studies: the design, contribution and interpretation of studies validating the inactivation and removal of viruses”, CPMP/BWP/268/95. [online] Available at: http://wwweurope.eu/docs/en_GB/document_library/Scientific_guideline wc 50000 3684.pdf2009/09/.
408 Burnouf, T. et al. (2005). “Place of Nanofiltration for Assuring Viral Safety of Biologicals”. Current Nanoscience, 1(3), 189-201.
409 Terpstra, F. G. et al. (2006). “Viral safety of Nanogam, a new 15 nm-filtered liquid immunoglobulin product”. Vox Sanguinis, 90(1), 21–32.
410 Roth, N. J. et al. (2020). “Nanofiltration as a robust method contributing to viral safety of plasma-derived therapeutics: 20 years’ experience of the plasma protein manufacturers”. Transfusion, 60(11), 2661–2674.
411 Roth, N. J. et al. (2020). “Nanofiltration as a robust method contributing to viral safety of plasma-derived therapeutics: 20 years’ experience of the plasma protein manufacturers”. Transfusion, 60(11), 2661–2674.
412 Kempf, C. et al. (1991). “Inactivation of human immunodeficiency virus (HIV) by low pH and pepsin”. Journal of Acquired Immune Deficiency Syndromes, 4(8), 828–830.
413 Omar, A. et al. (1996). “Virus inactivation by pepsin treatment at pH 4 of IgG solutions: factors affecting the rate of virus inactivation”. Transfusion, 36(10), 866–872.
414 Burnouf, T. et al. (2005). “Place of Nanofiltration for Assuring Viral Safety of Biologicals”. Current Nanoscience, 1(3), 189-201.
415 Boschetti, N. et al. (2005). “Virus safety of intravenous immunoglobulin: future challenges”. Clinical Reviews in Allergy and Immunology, 29(3), 333–344.
416 Bos, O.J. et al. (1998). “Virus validation of pH 4-treated human immunoglobulin products produced by the Cohn fractionation process”. Biologicals, 26, 267-276.
417 Louie, R.E., Galloway, C.J. & Dumas, M.I. (1994). “Inactivation of hepatitis C in low pH intravenous immunoglobulin”. Biologicals, 22, 13-19.
418 Cai, K. et al. (2005). “Ensuring the biologic safety of plasma-derived therapeutic proteins: detection, inactivation, and removal of pathogens”. BioDrugs: Clinical Immunotherapeutics, Biopharmaceuticals and Gene Therapy, 19(2), 79–96.
419 Lebing, W. et al. (2003). “Properties of a new intravenous immunoglobulin (IGIV-C, 10%) produced by virus inactivation with caprylate and column chromatography”. Vox Sanguinis, 84, 193-201.
420 Morfini, M. et al.(2013). “Clinical use of Factor VIII and Factor IX concentrates”. Blood Transfusion, 11 Suppl 4 (Suppl 4), s55–s63.
421 Hrinda, M.E., D’alisa, R. & Crissman Tarr, G. (1990). “Patent CA-2034489-C: Methods for the inactivation of viruses in viral-contaminated pharmaceutical compositions”. National Center for Biotechnology Information, PubChem Patent Summary for CA-2034489-C. [online] Available at:https://pubchem.ncbi.nlm.nih.gov/patent/CA-2034489-C.
422 Francisco, O.A. et al. (2019). “Do soft anions promote protein denaturation through binding interactions? A case study using ribonuclease A”. RSC Advances, 9, 3416-3428.
423 European Medicines Agency. (2018). “Guideline on the clinical investigation of recombinant and human plasma-derived Factor VIII products”. EMA/CHMP/BPWP/144533/2009 rev.2. [online] Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-recombinant-human-plasma-derived-factor-viii-products-revision-2_en.pdf.
424 Swiech, K., Picanço-Castro, V., & Covas, D. T. (2017). “Production of recombinant coagulation factors: Are humans the best host cells?”. Bioengineered, 8(5), 462–470.
425 Wurm, F.M. (2004). “Production of recombinant protein therapeutics in cultivated mammalian cells”. Nature Biotechnology, 22(11), 1393-1398.
426 Klein, U. (1991). “Production and characterization of recombinant Factor VIII”. Seminars in Hematology, 28(2 Suppl 1), 17–21.
427 Boedeker, B. G. (1992). “The manufacturing of the recombinant Factor VIII, Kogenate”. Transfusion Medicine Reviews, 6(4), 256–260.
428 Gomperts, E., Lundblad, R., & Adamson, R. (1992). “The manufacturing process of recombinant Factor VIII, recombinate”. Transfusion Medicine Reviews, 6(4), 247–251.
429 Gitschier, J. et al. (1984). “Characterization of the human Factor VIII gene”. Nature, 312(5992), 326–330.
430 Wood, W. I. et al. (1984). “Expression of active human Factor VIII from recombinant DNA clones”. Nature, 312(5992), 330–337.
431 Vehar, G. A. et al. (1984). “Structure of human Factor VIII”. Nature, 312(5992), 337–342.
432 Toole, J. J. et al. (1984). “Molecular cloning of a cDNA encoding human antihaemophilic factor”. Nature, 312(5992), 342–347.
433 White, G. C. et al. (1989). “Use of recombinant antihemophilic factor in the treatment of two patients with classic hemophilia”. The New England Journal of Medicine, 320(3), 166–170.
434 White, G. C. et al. (1997). “A multicenter study of recombinant Factor VIII (Recombinate) in previously treated patients with hemophilia A. The Recombinate Previously Treated Patient Study Group”. Thrombosis and Haemostasis, 77(4), 660–667.
435 Schwartz, R. S. et al. (1990). “Human recombinant DNA-derived antihemophilic factor (Factor VIII) in the treatment of hemophilia A. recombinant Factor VIII Study Group”. The New England Journal of Medicine, 323(26), 1800–1805.
436 Lusher, J. M. et al. (1993). “Recombinant Factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. Kogenate Previously Untreated Patient Study Group”. The New England Journal of Medicine, 328(7), 453–459.
437 Bray, G. L. et al. (1994). “A multicenter study of recombinant Factor VIII (recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. The Recombinate Study Group”. Blood, 83(9), 2428–2435.
438 Gruppo, R. et al. (1998). “Safety and immunogenicity of recombinant Factor VIII (Recombinate) in previously untreated patients: a 7.3 year update”. Haemophilia, 4(228). Abstract 291, XXIII Congress of the WFH, The Hague.
439 Lusher J. M. (1994). “Summary of clinical experience with recombinant Factor VIII products--Kogenate”. Annals of Hematology, 68(Suppl 3), S3–S6.
440 Lusher, J. et al. (2004). . “Human recombinant DNA-derived antihemophilic factor in the treatment of previously untreated patients with hemophilia A: final report on a hallmark clinical investigation”. Journal of Thrombosis and Haemostasis, 2(4), 574–583.
441 Ehrenforth, S. et al. (1992). Incidence of development of Factor VIII and Factor IX inhibitors in haemophiliacs. The Lancet, 339(8793), 594–598.
442 Addiego, J. et al. (1993). Frequency of inhibitor development in haemophiliacs treated with low-purity Factor VIII. The Lancet, 342(8869), 462–464.
443 Kreuz, W. et al. (2005). “Full-length sucrose-formulated recombinant Factor VIII for treatment of previously untreated or minimally treated young children with severe haemophilia A: results of an international clinical investigation”. Thrombosis and Haemostasis, 93(3), 457–467.
444 Blanchette, V. S. et al. (2008). “Plasma and albumin-free recombinant Factor VIII: pharmacokinetics, efficacy and safety in previously treated pediatric patients”. Journal of Thrombosis and Haemostasis, 6(8), 1319–1326.
445 Sandberg, H. et al. (2001). “Structural and functional characteristics of the B-domain-deleted recombinant Factor VIII protein, r-VIII SQ”. Journal of Thrombosis and Haemostasis, 85(1), 93–100.
446 Mikaelsson, M. et al. (1993). “Manufacturing and characterization of a new B-domain deleted recombinant Factor VIII, r-VIII SQ”. Thrombosis and Haemostasis. 69, 1205.
447 Recht, M. et al. (2009). “Clinical evaluation of moroctocog alfa (AF-CC), a new generation of B-domain deleted recombinant Factor VIII (BDDrFVIII) for treatment of haemophilia A: demonstration of safety, efficacy, and pharmacokinetic equivalence to full-length recombinant Factor VIII”. Haemophilia, 15(4), 869–880.
448 Lusher, J. M. (2004). “Is the incidence and prevalence of inhibitors greater with recombinant products? No.” Journal of Thrombosis and Haemostasis, 2(6), 863–865.
449 Larson, P. et al. (2004). “IgG formation to mammalian proteins in hemophilia A patients following treatment with a new recombinant human Factor VIII”. Journal of Thrombosis and Haemostasis, 2(6), 1011–1012.
Each contributing group member confirms that he or she understands his or her duty to provide independent evidence and has complied with that duty.
All contributing group members confirm that in respect of those parts of the report to which they have contributed:
Dr Ruth Laub is an independent consultant specialising in plasma derivative production and safety. She has twenty years’ experience as Associate Production Director and Research & Development Manager at the Red Cross Central Fractionation Department, Belgium. There she developed an original, semi-automated FVIII purification chain to produce VIII SD concentrates from cryoprecipitate. She holds a PhD from the Free University of Brussels and her research, at the de Duve Institute, Brussels (Catholic University of Leuven) involved neutral proteases, ferritin and siderophores. She focused later on FVIII epitopes, autoantibodies and their inhibitors, specific immunoglobulins, pollutant and virus removal from plasma derivatives, and virus inactivation techniques. She has served on the Board of Directors of Fonds de Biotechnologie – Biotechnologie Fonds, BIO.BE (Essencia), and Belgian Biotechnology Association. She has been a member of the International Society of Blood Transfusion, and the World Health Organisation International Working Group on Standardisation of Gene Amplification Technologies for the Virological Testing of Blood Products (SoGAT).
Dr Paul Strengers is an independent consultant specialising in blood and plasma strategies after a long-standing career in blood transfusion, plasma manufacturing and plasma derived medicinal products with research in clinical transfusion medicine and sciences. He holds a Medical Degree from the State University of Leiden, Netherlands, and was a Fellow of the Faculty of Pharmaceutical Medicine of the Royal Colleges of Physicians, UK. He has been Medical Director of the Blood Bank of the Central Laboratory of the Netherlands Red Cross Blood Transfusion Service; Director of Medical Affairs and Product Development at Sanquin Blood Supply Foundation/Sanquin Plasma Products and Medical Director of CAF-DCF/Plasma Industries Belgium, Brussels, Belgium. He has held memberships of many national and international organisations and committees on blood banks, donorship, transfusion and plasma fractionation, including the Netherlands Society of Blood Transfusion, International Association for Biologicals and Netherlands Society of Pharmaceutical Sciences. He was President of the International Haemovigilance Network, Secretary-General of the International Society of Blood Transfusion, and President of the International Plasma and Fractionation Association. He is a Member of the Expert Committee on Biological Standardisation (section Blood) of the World Health Organisation.