January 2020
with addendum
![]()
© Crown copyright 2020
This publication is licensed under the terms of the Open Government Licence v3.0 except where otherwise stated. To view this licence, visit nationalarchives.gov.uk/doc/open-government-licence/version/3
Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned.
This publication is available at www.infectedbloodinquiry.org.uk
Any enquiries regarding this publication should be sent to us [email protected]
01/20
Printed on paper containing 75% recycled fibre content minimum
Printed in the UK by the APS Group
Explanatory Note
Please note that the paragraph numbers below relate to numbered questions in the Letter of Instruction which was sent to the Hepatitis Expert Group by the Infected Blood Inquiry
Hepatitis refers to inflammation of the liver, regardless of the cause. A diagnosis of hepatitis is usually made by detection of biochemical abnormalities in the blood, in particular measuring enzymes released from liver cells (hepatocytes) damaged by inflammation. Two enzymes are key to liver function testing; alanine aminotransaminase (ALT) and aspartate aminotransferase (AST). Hepatitis is usually diagnosed when one (or more) of these enzymes is raised above normal ranges in the context of other results. Definitions of normal range vary with those for ALT and AST most commonly quoted as 5-40 IU/l. In recent years recommendations have emerged suggesting these ranges are too high and may miss individuals with mild hepatitis. Current US guidelines recommend a normal range for ALT of 29-33 IU/l for men and 19-25 IU/l for women.1
There is a wide range of causes of hepatitis, including infection (most commonly viruses, but also bacteria and parasites), medication and a high alcohol intake. Fatty liver disease associated with obesity (referred to as non-alcoholic fatty liver disease, NAFLD) is an increasingly important cause of hepatitis. Other rarer causes include metabolic disorders (such as haemochromatosis) and autoimmune disease. Routine testing for the cause of hepatitis will usually involve a panel of blood tests, imaging of the liver (ultrasound) and, in some cases, a liver biopsy.
Hepatitis B virus (HBV) and hepatitis C virus (HCV) are the most important causes of viral hepatitis globally. Each can range in severity from very mild, where an individual has no symptoms and no long-term consequences, to being so severe that the liver can no longer carry out its essential functions and fails with a high risk of death, sometimes necessitating transplantation (see Q15.11). The key concern with long term HBV or HCV infection is progressive scarring of the liver (fibrosis, leading to cirrhosis) and an increased risk of liver cancer (hepatocellular carcinoma, HCC).
The key viruses transmitted via blood are hepatitis B (HBV) and hepatitis C (HCV). Hepatitis D (delta) virus can only infect individuals already infected with HBV. Other major causes of viral hepatitis (hepatitis A and E) are primarily transmitted via contaminated food or water, rather than blood.
Together, HBV and HCV viruses are amongst the leading causes of mortality globally, responsible for more deaths each year than HIV or malaria.2
Hepatitis B is a DNA (deoxyribonucleic acid) virus. Despite the availability of a highly effective vaccine, an estimated 3 million individuals are infected globally each year. The majority of these infections are in infants and children in sub-Saharan Africa.
There are eight recognized HBV genotypes (A to H) amongst an estimated 257 million individuals living with chronic HBV infection worldwide. Genotype A is found mainly in North America, northern Europe, India and Africa, genotypes B and C are prevalent in Asia and genotype D is more common in southern Europe, the Middle East and India. In a large UK study of hepatitis B, genotype D was most common (31%) followed by A, C, B and E (20%, 20%, 19% and 9%, respectively).3
The clinical relevance of HBV genotype is relatively limited, in contrast to hepatitis C (see below). Most information on the clinical significance of different HBV genotypes has been derived from Asian studies of chronic infection with HBV genotypes B and C. For example, the prevalence of highly replicating infection (Hepatitis B “e” antigen, HBeAg, positive) appears higher in patients with genotype C rather than genotype B. There is limited information on the clinical course of patients with HBV genotypes other than B or C.4 A few studies have suggested that HBV genotype D infection is more likely to be associated with fulminant hepatitis and that HBV genotype A infection is more likely to progress to chronic infection.
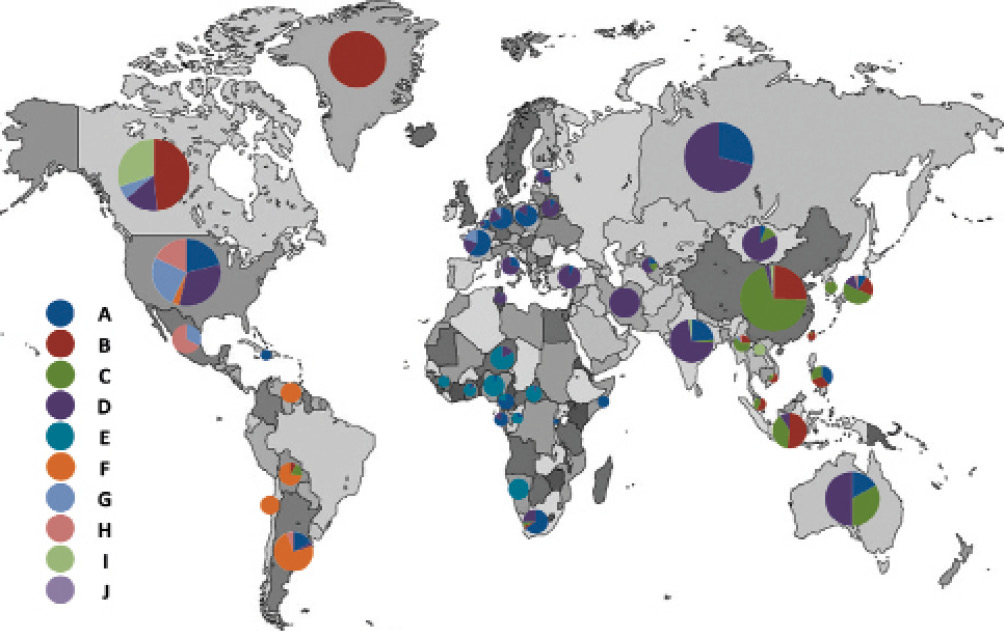
Figure 15.2a Global distribution of hepatitis B genotypes (reproduced from Shi et al5)
In contrast to hepatitis B, hepatitis C is an RNA (ribonucleic acid) virus. Eight genotypes of HCV, and more than 80 subtypes, have been described.6 Genotype 1 is most common globally (45% of isolates); genotype 3 is most common in India and the Far East (30% of isolates globally). Genotypes 2, 4 and 6 account for 23% of global infection. Genotype 4 is most common in Africa and the Middle East, particularly in Egypt, and genotype 6 is most frequent in Hong Kong, Vietnam and Laos. Genotype 5 is rare, but found in South Africa (see Figure 15.2b).
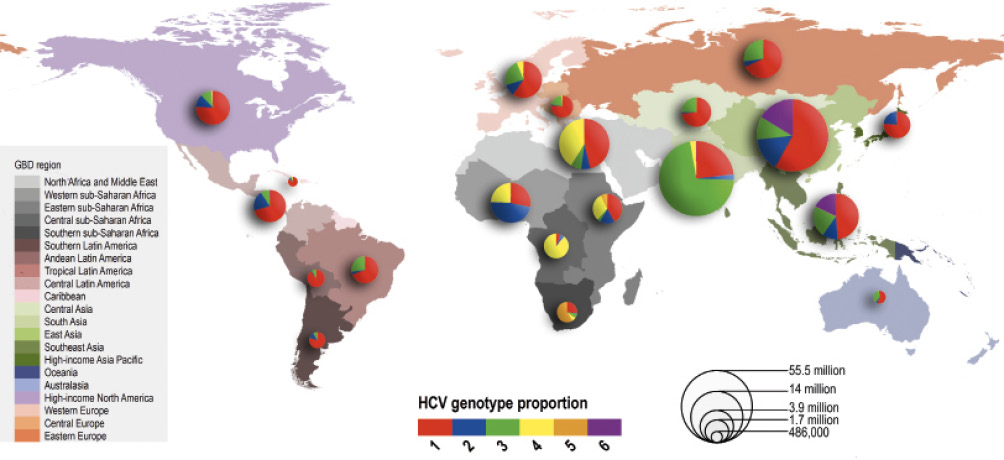
Figure 15.2b Relative genotype distribution of hepatitis C amongst estimated 71 million people living with the virus globally (from Messina et al 7)
In the UK, genotype 1 and 3 each account for approximately 40% of infections respectively, the relatively high proportion of genotype 3 reflecting a larger population migrating from Southeast Asia than from other European countries.
Viral genotypes are important for treatment choice and duration, most notably when treatment was based on interferon. Even in the current era of new treatment (see Q15.13) genotype is often important for the choice of therapy, though with the development of treatments active across all main genotypes (pan-genotypic), genotypes are less important in decision making than they were.
Hepatitis D virus (HDV), also known as hepatitis delta virus, is a defective RNA virus. Although HDV can replicate, it requires the presence of the HBV virus (HBsAg) to assemble and secrete new viruses. Thus, all individuals with HDV are also co-infected with HBV. Patients may be either simultaneously infected with HBV and HDV, or an HBV carrier may be superinfected with HDV.
Based on an estimate of 257 million HBsAg positive individuals globally of whom 5-10% are co-infected with HDV, there are 12-25 million individuals living with HBV/HDV co-infection. A high prevalence of HDV is found in eastern Europe, Turkey Russia and former Soviet states, as well as Pakistan, Mongolia, the northwest of South America and several countries in Africa, including Somalia, Kenya, Benin and Gabon.8 In the UK the prevalence of HDV is estimated at approximately 3% of those infected with hepatitis B and there is limited data on genotypes.
Eight genotypes of HDV have been described. Genotype 1 is widely distributed across the globe, genotype 3 is concentrated in South America, genotypes 2 and 4 are mainly found in Southeast Asia (with some genotype 2 found in Egypt and Russia), and genotypes 5-8 in Africa. There is limited data allowing meaningful comparison of the clinical relevance of genotypes, though genotype 3 in the Amazonian region has been associated with a poor prognosis.9
It is not clear when hepatitis B first emerged, but it has been found in human remains up to 4500 years old. Although hepatitis B is a DNA (deoxyribonucleic acid) virus it replicates using an enzyme known as an RNA (ribonucleic acid) polymerase which lacks proof-reading ability, thereby introducing genetic variation, especially under immune pressure from the host. This results in the coexistence of genetically distinct viral species in infected individuals, also called viral quasispecies, which evolve depending on the pressure from the host environment. It is likely that (as with hepatitis C) the virus has spread geographically in last 100 years due to infection via medical transmission, infected blood products, injecting drug use and other routes, including human migration. In recent years, migrants and refugees from outside Europe, with high rates of hepatitis B infection, are changing the prevalence and incidence of hepatitis B in Europe, in areas of low endemicity (eg Italy, Germany). In addition, in contrast to hepatitis C, hepatitis B is readily spread via sexual transmission (see Q15.5).
As discussed in Q15.2, although viral genotypes are less important clinically than for hepatitis C, there are pathogenic differences between the HBV genotypes which partially explain disease intensity, progression to liver cirrhosis, and hepatocellular carcinoma (HCC).
Using a technique known as the phylogenetic clock (looking at the rate of molecular change to allow tracing back to a time point when species are predicted to have diverged), it is estimated that hepatitis C (HCV) first emerged over 3,000 years ago. HCV is an RNA (ribonucleic acid) virus and, in common with all RNA viruses, it circulates in a host as a complex, genetically related, but heterogenous population referred to as a quasispecies. This is principally due to the fact that the enzyme (known as an RNA polymerase) required for virus multiplication lacks proof-reading ability (and thus introduces mutations into the virus), but also because of immune pressure exerted by the host. In the last 100 years or so, a few genotypes (specifically 1a, 1b, 2a, 3a: ‘epidemic subtypes’) have spread rapidly around the world and become predominant. These endemic subtypes have spread via medical transmission, infected blood products, injecting drug use and other routes, including human migration. Other less common types (‘endemic strains’) have circulated for long periods of time in areas restricted by geography.
It is not clear whether certain genotypes are more likely to be associated with spontaneous clearance of the virus without treatment, as the genetic background of the host is also important, and most of the studies that look at genotype and viral clearance are based on outbreaks which occur with a single genotype and often in a genetically similar population. However, the natural history of infection is influenced by virus genotype; patients with genotype 3 tend to progress more rapidly to fibrosis and cirrhosis, with a higher prevalence of severe steatosis (fatty liver) and a higher incidence of hepatocellular carcinoma (liver cancer) – reviewed in Shahnazarian et al.74 HCV genotype predicts successful treatment – this is discussed in section 15.14.
With advent of DAAs, there is greater interest in naturally occurring variation in the virus and how that impacts treatment outcome (so-called resistance associated substitutions, or RASs). These viral variants are circulating in the population and may reduce the rates of curative treatment. Increasingly, clinical important subtypes are being recognised (for example genotype 4r, which is relatively common in East Africa).75
For the avoidance of doubt, the information set out below describes current practice in the UK. Blood for transfusion in the UK is collected from volunteer donors who have met donor selection criteria. These criteria form part of the transfusion guidelines for the UK, published online by the Joint United Kingdom Blood Transfusion and Tissue Transplantation Services Professional Advisory Committee (JPAC).76
At the time of each blood donation, samples are taken to be tested to determine the donation’s blood group. Microbiological testing is also performed for hepatitis B, hepatitis C, hepatitis E, HIV, HTLV and syphilis. Further microbiological testing may be performed if indicated, for example testing for malaria or West Nile Virus when donors have a relevant travel history. Blood components are not released for transfusion until the results of these tests are known and confirm the donation to be free of infection.
A standard donation is around 475ml of whole blood. This whole blood is further processed into blood components. Red cells are resuspended in additive solution to optimise conditions for red cell survival prior to transfusion. Plasma is frozen promptly following donation as ‘fresh frozen plasma’ (FFP), which preserves the concentration of blood clotting factors present. FFP can be further processed into cryoprecipitate, a plasma fraction rich in fibrinogen, von Willebrand factor and Factor VIII. Cryoprecipitate was initially manufactured as a treatment for haemophilia A, but is no longer used in this context due to the availability of recombinant clotting factor concentrates. Leucocyte depletion is performed on all blood components to remove white cells. Platelets for transfusion can be derived from the buffy coat of whole blood donations or collected by apheresis, a process by which platelets specifically are collected from a donor.
Plasma derivatives (also known as blood products) differ from blood components in that they are manufactured from the pooled plasma of many (sometimes thousands of) donations, rather than a single donation. These large plasma pools undergo fractionation to collect the plasma constituent of interest, and undergo pathogen inactivation steps, for example solvent/detergent treatment or methylene blue treatment, to reduce the risk of transfusion transmitted infection. Examples of such plasma derivatives include clotting factor concentrates, immunoglobulin and albumin. It should be noted that recombinant (non-plasma derived) clotting factor concentrates are the first choice treatment for inherited clotting factor deficiencies, including haemophilia A and haemophilia B, and will be used in preference to plasma derived products. Since 1999, plasma derivatives used in the UK have been sourced from outside the UK as a variant Creutzfeldt-Jakob Disease (vCJD) risk reduction measure.
Ensuring blood components and blood products are only transfused appropriately is a key step in preventing transfusion transmitted infection. It is unusual now for whole blood to be transfused to a patient; rather blood components as described above are transfused as indicated. National guidelines outlining the appropriate use of blood and blood components have been published by NICE65 and the British Society for Haematology (BSH).77 Broadly speaking, red cells are transfused to treat anaemia which may be caused by many different conditions: haematological malignancies, as a result of cytotoxic chemotherapy, as a result of an inherited condition like sickle cell disease or thalassaemia or kidney disease. The majority of platelet transfusions are administered to patients with haematological malignancies and those undergoing chemotherapy, both of which can impair normal platelet production leaving the patient at risk of bleeding. FFP may be transfused to correct coagulation abnormalities before surgery. In the context of acute blood loss, patients will be transfused a combination of blood components aiming to maintain the patient’s circulation and to prevent abnormalities of blood coagulation which can result from significant blood loss.
The Blood Safety and Quality Regulations 200578 and JPAC guidelines76 mandate that blood donations are screened for the presence of hepatitis infections using serological tests and nucleic acid testing (see Q15.6). The combination of these two technologies means that the overwhelming majority of infected blood donations are detected and discarded before any transfusion takes place. The small risk remains, however, that a donation with very low levels of viral nucleic acid present may pass these screening tests, leading to potential for transfusion of an infected component. This scenario is most likely to occur during the early stages of viral infection in the donor, when viral load is too low to be detected by nucleic acid (either RNA or DNA) testing and serology tests are not yet positive: the so-called window period. With advances in testing technologies the window periods for hepatitis viruses has reduced over time. The risk of standard testing by the blood services missing an occult infection is highest for hepatitis B due to its relatively long window period and is estimated at approximately 1.04 per million donations.79 This translates to one such donation being missed by screening every 6 months. The much shorter window period for hepatitis C makes failing to identify an infection much less likely and it is estimated a window period donation may be missed only once every 90 years.79
It must also be noted that risk of failure to detect an infection by testing does not equate to risk of transmission of infection and that infection rates are lower, because these calculations do not take into account that some blood packs do not go on to be transfused nor do they consider the susceptibility of a given patient to the infection. Data from SHOT80 demonstrate there has not been a confirmed case of transfusion transmitted HCV in the UK since 1997, and one confirmed and two probable cases of transfusion transmitted HBV in the last 10 years.
When considering the risk of transmission of a hepatitis virus posed by a specific infected component or product, various factors must be considered. Firstly, the type of component; it is considered that plasma products (FFP, cryoprecipitate, plasma derivatives) pose a higher risk as the viral titre of these is higher than of a cellular blood component; red cell and platelet components contain only a small volume of residual plasma following manufacture. Secondly, the volume of transfusion is also important, with recipients of large volume transfusions of infected product receiving a higher absolute viral load. This will vary with the component or product being used as well as the indication for transfusion. The viral titre of the infected component is also important with those with the highest viraemic titres posing the greatest risk. In the context of contemporary donor screening practices using NAT based assays it is estimated that a whole blood donation which screens negative for hepatitis B infection could contain up to 384 IU/mL of viral genome and for hepatitis C up to 480 IU/mL.81 Recent data suggest the minimum infectious dose for hepatitis B to be of the order of 3IU of viral DNA, indicating that window period donations are capable of transmitting infection.82 Finally, the recipient may be more or less susceptible to infection, with immunosuppressed individuals at greater risk.
As discussed above, plasma derivatives are subject to pathogen inactivation steps to eradicate viruses which may be present in the source plasma. Solvent/detergent treatment employed in the manufacture of such plasma derivatives inactivates lipid-envelope viruses, which include the hepatitis B and hepatitis C viruses, minimising the risk of transmission of these viruses through transfusion of these blood products.
Transmission of both HBV and HCV can occur from mother to child during pregnancy or around the time of delivery and has been reviewed in detail.83 Globally, vertical transmission remains the main route of infection for HBV. For both HBV and HCV higher levels of virus in the mother and the presence of HIV are both strongly associated with higher rates of transmission.83
Up to 40% of transmission of HBV is before the onset of labour. Mechanisms include leakage across the placenta (particularly if the placenta is damaged), infection of the placenta, transfer in infected white blood cells which traffic between mother and foetus, or direct infection of sperm/eggs. Transmission at delivery can result from trauma, or contact of the neonate with the lining of the vagina. Breastfeeding does not transmit HBV (unless there is clear breakage to the skin/bleeding).
Approximately 30% of transmission of HCV occurs before labour. As with HBV, infection or damage to the placenta can allow infection of the foetus. A specific transport mechanism has been identified that can carry HCV across the placenta. At delivery any traumatic contact can allow the virus to pass to the neonate. Like HBV, breastfeeding does not transmit HCV.
Studies investigating rates of vertical transmission for HBV and HCV have produced widely varying estimates (on average 1-28% for HBV and 3-15% for HCV82 but potentially much higher in selected individuals, such as those with a very high viral load), but in general vertical transmission is considered more likely in the setting of HBV (at approximately 5%)84 compared to <5% for HCV.85 In areas of high prevalence transmission between children (horizontal transmission) remains common. Infants infected in early life are much more likely to develop long-term (chronic) infection than adults.
HBV and HCV are both transmitted through contaminated needles and syringes that may be used for medical reasons, recreational drug use, tattooing or piercings. This is a much more common route of transmission for HCV than HBV, though unsafe injection remains an important source of adult infection with HBV. The reuse or sharing of needles is a major factor in transmission, and provision of disposable, clean needles is one of the key public health measures able to reduce transmission.
HBV is present in vaginal and seminal fluid, and rates of transmission and sexual transmission is estimated to be 50-100 times higher than for HCV. Sexual transmission to unvaccinated partners of individuals with chronic HBV is common and up to 40% of partners may become infected.86,87 Detailed estimates of the rates of transmission are less relevant in the current era where vaccination coverage has improved significantly, but sexual transmission is a common route of transmission in adults, particularly between men who have sex with men (MSM).
Although HCV is detectable in seminal and vaginal fluid, sexual transmission of HCV between heterosexual couples is rare, estimated at 0.07%/year or 1 in 190,000 occurrences of intercourse.88 One reason for this difference may be lower levels of virus in genital secretions compared to HBV. Sexual transmission of HCV between MSM is far more common, particularly since the advent of highly effective treatment for HIV and more widespread use of pre-exposure prophylaxis (PREP).
Serology: The majority of HBV and HCV infections are initially diagnosed using enzyme immunoassays (EIA), which are widely used for screening large groups of patients, as they are relatively inexpensive and can be highly automated. These assays are also known as serological assays as they are usually performed on blood serum or plasma samples, but may also be performed on capillary/venous whole blood and oral fluid. Other serological assay formats include rapid diagnostic tests (RDTs) and chemiluminescence immunoassays (CLIAs). The most recent assay format is the Chemiluminescent Microparticle Immuno Assay (CMIA).
Most serological assays are designed to detect specific antibodies that are produced in response to infection rather than directly detecting the virus. This is true for HBV (anti-HBcoreAb) and HCV (anti-HCV), but in the case of hepatitis assays have also been designed to look directly at protein components (known as antigens as they provoke an antibody response, for example HBsAg, HCVcoreAg). The principle of any EIA is that an antigen or antibody of interest will specifically bind to the complementary antigen/antibody (see Figure 15.6a) which is immobilised on a solid surface, for example a 96-well plate or a bead. If the target of interest is present in the patient’s serum it will specifically bind to the immobilised complementary antigen/antibody. This binding can then be detected by using a secondary antibody which specifically binds to the antigen or antibody of interest and which has an enzyme attached to it. A chemical is added to be converted by the enzyme into a colour or a fluorescent signal. The amount of signal produced can be quantified and gives a measure of the amount of antigen or antibody in the patient’s sample. Between each step the plate is washed with a mild detergent to remove any antigens or antibodies that are non-specifically bound. This is known as a ‘sandwich’ or indirect EIA.
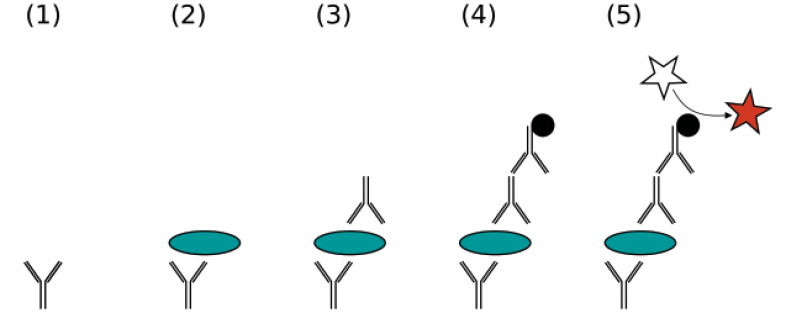
Figure 15.6a A sandwich ELISA (1) Plate is coated with a capture antibody; (2) sample is added and any antigen present binds to capture antibody; (3) detecting antibody is added and binds to antigen; (4) enzyme-linked secondary antibody is added and binds to detecting antibody; (5) substrate is added and is converted by enzyme to detectable form.
Attribution Jeffrey M. Vinocur, https://commons.wikimedia.org/wiki/File:ELISA-sandwich.svg
Nucleic acid testing: The other key group of diagnostic assays for HBV and HCV are molecular assays or nucleic acid testing (NAT), for example polymerase chain reaction (PCR) or nucleic acid sequence-based amplification (NASBA), which can detect very small quantities of viral nucleic acid (RNA or DNA). As well as detecting the presence of the virus, these assays provide quantification of the virus in either virus copies/ml or, more recently, as standardised international units (IU)/ml). These quantified assays are generally referred to as measures of “viral load”. The assays detect DNA or RNA through targeting a specific segment of the virus, which is then amplified. The amplification step enables the detection of low levels of the virus in the original specimen which might not otherwise have been detectable. Laboratory-based technologies for NAT traditionally require sophisticated equipment, rigorous laboratory conditions and specimen collection, and highly trained staff who can perform precision steps and avoid contamination. More recent technological innovations allow the use of these assays at the point of care (such as prisons or remote rural locations), although they still require trained staff and specialist equipment, and have a considerable cost.
NAT technologies are typically used to detect the presence of the virus, determine if the infection is active and if the individual would benefit from antiviral treatment. NAT technologies are also used to determine when antiviral treatment should be discontinued (due to non-response or resistance) or to confirm virological cure (HCV) or effective suppression (HBV). These assays are high cost compared with serological assays. Specialist virology laboratories were providing non-commercialised NAT based assays (qualitative) from the early 1990s, often developed ‘in-house’, with more automated and quantitative assays becoming available in the second half of that decade. More sensitive and highly automated assays, using real-time PCR technology, have been used routinely since the mid to late 2000s.
Measures of Test Performance: Key attributes of any diagnostic test are the sensitivity (that is, the extent to which a test correctly identifies those with the disease (true positive rate)) and specificity (that is the ability of the test to identify those without the disease (true negative rate)). A test with 100% sensitivity correctly identifies all those with the condition of interest; anything less than a sensitivity of 100% will mean that an individual may go undetected (false negative). A test with 100% specificity correctly identifies all those without the condition of interest; anything less than 100% will mean that an individual may be incorrectly diagnosed as being test positive (false positive). In general, screening tests designed for the diagnosis of HBV and HCV have a high sensitivity so that individuals with the diagnosis are not missed, which is important when such tests are used in the setting of blood donor screening. Before a confirmed diagnosis is made, further confirmatory tests with a high specificity are required.
However, sensitivity and specificity of a test only describe how well the test performs against the gold-standard test for that disease and will depend on the population it is being tested on. For example, when a test is applied to a population with a low prevalence of the infection/disease in question, unless the test has 100% specificity, the number of false-positive results will be higher than when testing a population with a high prevalence.
Hepatitis B virus first came into view in 1965 with the discovery of the so-called Australia antigen (AuAg) by screening blood samples from multiply transfused individuals.10 The association of this antigen with multiple blood transfusions, and with cases of seroconversion (new detection of the antigen) in association with the development of hepatitis, led to the association of this antigen with an infectious hepatitis, and ultimately to type B viral hepatitis.89 Later, in London the electron microscopy work of David Dane and June Almeida showed that AuAg was in fact the surface antigen of the viral envelope and was named hepatitis B surface antigen (HBsAg).
Diagnostic assays for HBV are generally performed on venous blood samples in laboratories designated for testing for infectious diseases. Serological assays are the primary diagnostic method. The serological definition of hepatitis B infection is the presence of HBsAg in the blood. Chronic hepatitis B infection is defined by the continued presence of HBsAg in the blood for longer than six months. Standardised enzyme-linked immunoassays (EIA) for hepatitis B have been designed to detect multiple hepatitis B antigens and antibodies in order to diagnose and characterise the infection. HBsAg, anti-HB core (anti-HBcAb), and anti-hepatitis B e antigen (anti-HBeAb) and hepatitis B e antigen (HBeAg) are detected by standardised enzyme-linked immunoassays (EIA). HBsAg is generally used as the screening assay for hepatitis B infection; in low prevalence populations false positive assay reactivity can occur. A neutralisation assay can be used to confirm the presence of HBsAg in human serum or plasma. A specific anti-HB surface antibody (anti-HBs) is added to the specimen, and if this abolishes (neutralises) reactivity in the HBsAg assay in comparison with a control reaction, this confirms that the detected HBsAg is a true positive result. The presence of other serological markers, such as anti-HB core, provide further confirmation of hepatitis B infection.
The changes in the blood that occur during symptomatic acute hepatitis B infection have been well characterised and are shown in the figure below. HBsAg is the first marker to become detectable on serological testing, usually about six weeks after inoculation. The incubation period prior to the onset of clinical symptoms from inoculation is between 60 and 180 days. In the majority of adults with a competent immune system, HBsAg declines after one to five months and the antibody against it, anti-HB surface antibody (anti-HBs), appears. Anti-HBc is detectable around the onset of clinical symptoms, and is a reliable and persistent marker that an individual has ever had hepatitis B infection.
Historically, much has been made of a ‘window period’ when neither HBsAg nor anti-HBsAb is detectable, and anti-HBcAb can be the only detectable sign of hepatitis B infection unless anti-HBeAb is measured. However, with the current highly sensitive assays, this is rare and the window period has been shortened by the development of more sensitive tests. The viral protein ‘e’ antigen (HBeAg) becomes detectable around the same time as HBsAg and usually disappears around the time of peak symptoms with the development of anti-HBe antibody. The appearance of anti-HBs indicates resolution of HBV infection.
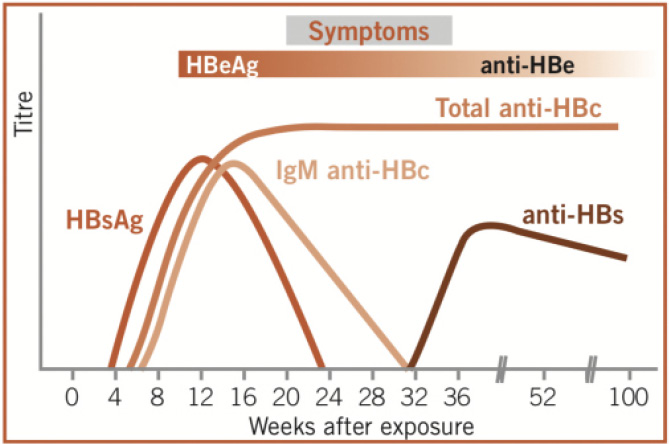
Figure 15.6b Detectable markers in the serum of patients with symptomatic acute hepatitis B infection
Between 5-10% of adults infected with HBV will develop a chronic carrier state, as shown in the figure below, with the continued presence of HBsAg, which is likely to persist potentially for life. The natural history of HBsAg loss is around 1% per year. The majority of individuals will seroconvert from HBeAg to anti-HBe during the course of their infection.
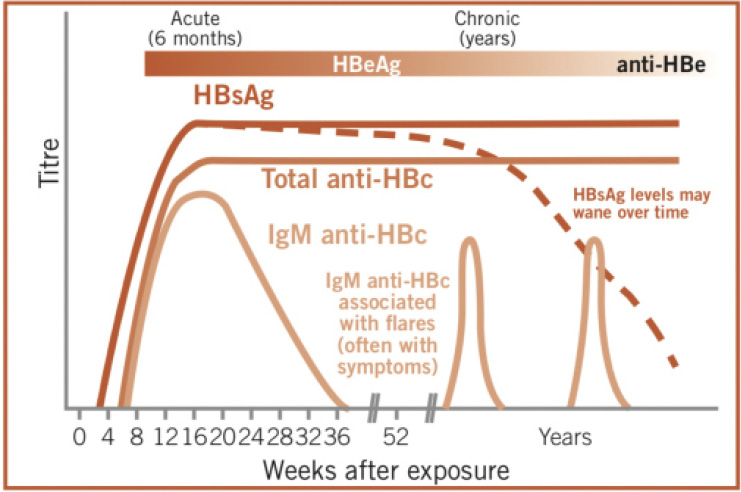
Figure 15.6c Detectable markers in the serum of patients with chronic hepatitis B infection
As can be inferred from the figures, because of the multiple serological tests available to diagnose hepatitis B, the combination of positive tests both confirms the diagnosis and provides an insight into the stage of disease. High levels of anti-HBc IgM are present during acute infection, but may remain detectable for up to 6 months – it can be used to differentiate between acute and chronic HBV infection, but its reappearance during “flares” in chronic HBV infection make it an unreliable indicator of recent primary HBV infection.
Molecular assays (NAT): are used to detect and quantitate HBV DNA. NAT testing for hepatitis B is not usually used for the primary diagnosis of HBV infection in clinical practice. However, it is used for the screening of blood products in order to close the short window period between the appearance of HBV DNA and HBsAg. The window period between HBV infection and detection of HBsAg is estimated to be around 38 days (depending on the analytical sensitivity of the assay and the period between the loss of HBsAg and the appearance of anti-HBs, when HBV DNA may still be detectable for a short period). There are also a small number of individuals with so-called ‘occult’ HBV infection (see Q15.17) who do not have detectable HBsAg, but have low level HBV DNA which could be transmitted via blood products.
Following diagnosis, the measurement of HBV DNA is now part of routine clinical assessment of patients with a diagnosis of hepatitis B infection, as levels help to guide the initiation of treatment in conjunction with other factors. HBV DNA levels also help determine if the anti-viral medication is effective and can be a guide to adherence. HBV DNA levels are an important part of assessment in pregnancy as they may guide treatment to prevent mother to child transmission.
It is important in patients newly diagnosed with hepatitis B to test for hepatitis D (HDV). Hepatitis D is a virus that only occurs in the presence of hepatitis B as it requires HBV in order to replicate (see Q15.2). Hepatitis D is diagnosed using serological methods (EIA). If anti-HDV antibodies are detected, NAT assays, to see if the individual has HDV viraemia, are recommended.
Decision to test for hepatitis: There are two approaches to the decision to test for hepatitis B infection: (i) because an individual presents with clinical features that could be consistent with hepatitis (see Q15.8), or (ii) because they meet an indication for screening, either as part of an at-risk population, or as a routine, such as in pregnancy or when making a blood donation.
All pregnant women in the UK are offered screening for hepatitis B and HIV as part of routine antenatal care. This programme accounted for 30.4% of all hepatitis B screening in participating sentinel centres in 2015.90 Genito-urinary medicine clinics also perform high numbers of hepatitis B and HIV tests as both infections are readily transmitted sexually, with general practitioners performing the largest proportion of screens for hepatitis B (33.1%).
NICE guidelines65 (UK based) recommend hepatitis B screening for the following groups on the basis that they are at increased risk of hepatitis B compared with the general UK population:
* It is no longer routine practice to screen for HBV in genito-urinary medicine clinics, in line with guidelines from the British Association of Sexual Health and HIV (BASHH) 2015 unless they trigger another high-risk criteria.
In addition to the groups at increased risk of HBV (key populations) defined by NICE, the WHO defines a number of testing approaches in the table below:
Table 15.6a Recommended approaches to the testing of HBV and HCV
|
Testing approaches for HBV and HCV |
|
|
Key populations |
Groups of people who due to specific high-risk behaviours, are at increased risk for HIV infection irrespective of the epidemic type or local context, but this may also apply to HBV and/or HCV infection. Key populations often have legal and social issues related to their behaviours that increase their vulnerability to HBV and HCV infection. These guidelines refer to the following groups as key populations: men who have sex with men (MSM); people who inject drugs (PWID); people in prisons and other closed settings; sex workers; and transgender people. |
|
Vulnerable populations |
Groups of people who are particularly vulnerable to HBV/HCV infection in certain situations or contexts. These guidelines refer to the following groups as vulnerable populations: migrant and mobile workers; and indigenous populations. |
|
General population testing |
This approach refers to routine testing throughout the entire population without attempting to identify high-risk behaviours or characteristics. It means that all members of the population should have potential access to the testing programme. This approach might be indicated for those countries with an intermediate or high HBV or HCV seroprevalence. |
|
“Birth cohort” testing |
This approach means routine testing among easily identified age or demographic groups (specific “birth cohorts”) known to have a high HCV prevalence due to past generalised exposures that have since been identified and removed. Most countries have at least some component of a “birth cohort” epidemic for HCV, but it may also be evident for HBV as a result of the introduction of HBV vaccination. |
|
Antenatal clinic testing |
This approach means routine testing of pregnant women, especially in settings where there is an intermediate or high seroprevalence, to identify women in need of antiviral treatment for their own health, and additional interventions to reduce mother-to-child transmission (MTCT). |
|
Community-based testing |
Includes using outreach (mobile) approaches in general and key populations; home-based testing (or door-to-door outreach); testing in workplaces, places of worship, parks, bars and other venues; in schools and other educational establishments; as well as through campaigns (screening for HIV or malaria alongside that for noncommunicable diseases such as diabetes and hypertension, for example). |
|
Facility-based testing |
Includes testing in primary care clinics, inpatient wards and outpatient clinics, including specialist dedicated clinics such as HIV, STI and TB clinics, in district, provincial or regional hospitals and their laboratories, and in private clinical services. |
In the mid-1970s it was clear that there were cases of hepatitis occurring post-transfusion that were attributable to neither hepatitis A nor hepatitis B, resulting in the term ‘non-A non-B hepatitis’ (NANBH). It was known that the agent causing NANBH was transmissible from studies in primates. HCV, the responsible virus, was found using a direct molecular biological approach in 1989. This led on to the development of a diagnostic test for HCV antibody for the first time using enzyme immunoassays (EIA), as discussed at the beginning of this section.
These assays quickly demonstrated that NANBH was responsible for most of parenterally (that is injection/infusion) transmitted cases of NANBH infection and that they could be used to detect and screen out infected donors from the blood supply.91 By 1991, most industrialised nations (including the UK) had implemented first-generation HCV-antibody testing in the blood donor population.91 The detection of hepatitis C antibody alone is not sufficient to make a diagnosis of active hepatitis C infection, as an individual infected with hepatitis C will usually remain antibody positive life-long after clearance of the virus (spontaneously or by successful treatment). Direct detection of the virus is required to confirm active infection, usually by molecular diagnosis (detection of HCV viral RNA by PCR testing), but also by detection of viral components (HCV core antigen) as detailed below.
Diagnostic assays for HCV are generally performed on venous blood samples, in laboratories designated for testing for infectious diseases. Diagnostic assays for HCV now include:
(i) The first serological assays to be developed were enzyme immunoassays (EIA) to detect anti-HCV immunoglobulin (IgG). Three generations of these assays have now been utilised, each with improved sensitivity compared with the previous generation. Some commercial companies now use a Chemiluminescence Immunoassay (CLIA) format: this is an antibody test similar to the EIA and, for the diagnosis of HCV, has similar sensitivity and specificity as the third-generation EIA (see below). These assays are employed as the initial screening assay for HCV infection. If a screening assay is positive a combination of additional assays will be used to determine if the assay reactivity represents a ‘true positive’ result and whether the individual has evidence of current infection or not (as evidenced by a positive HCV core antigen test and/or detectable HCV RNA). Historically, samples testing positive for HCV antibody may have been confirmed using a Recombinant Immunoblot Assay (RIBA) which identified specific HCV antibodies generated in response to HCV antigens, allowing differentiation between true and false positive antibody results. The HCV RIBA assay has now been discontinued as it is not as sensitive or specific as other HCV assays. It has been replaced by more sensitive molecular assays, which are also able to confirm that an individual has evidence of current infection.
The EIA format has also been adapted for point of care use in hard to reach patient groups, such as in drug and alcohol treatment centres.
HCV core antigen testing is now available using immunoassay formats. It is quantitative and has a high level of sensitivity and specificity in comparison with HCV RNA virus loads above 3,000iu/ml. In many diagnostic algorithms it is incorporated into confirmatory testing protocols as a marker of active HCV infection. It has the potential to close the diagnostic window, but is not generally used as the first line screening test in routine diagnostic laboratories.
(ii) Molecular diagnostic tests for HCV specifically detect HCV RNA which is only present in cases with active HCV infection. The process is commonly referred to as an HCV PCR (polymerase chain reaction) or HCV NAT. The HCV NAT becomes positive approximately 1 to 2 weeks after initial HCV infection and has become the gold standard supplemental test for patients who have a positive HCV EIA screening test. HCV NAT tests can be qualitative (a result is ‘detected’ or ‘not detected’) or quantitative. Quantitative assays now have a very high level of sensitivity, detecting levels down to as low as 10.5iu/ml depending on the assay used. Although quantitative assays are not generally licensed for diagnosis, they are often used for diagnosis in conjunction with antibody testing. HCV RNA testing is higher in cost and generally slower in turnaround time than serological based assays (including HCV antigen testing) as the tests are usually performed as a batch of several samples together. However, random access molecular assay technologies are becoming widely available, although at higher cost than a batched assay.
The figure below (Figure 15.6d) shows the typical time course post-infection of virological and immunological markers of HCV infection with self-resolving HCV infection.
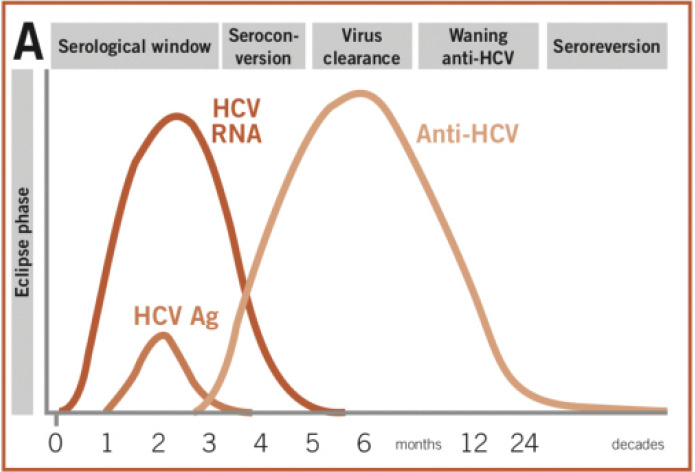
Figure 15.6d A: time course post-infection of virological and immunological markers in self-resolving HCV infection; detectable virus (HCV RNA and HCV antigen) peaks and starts to fall as antibodies (anti-HCV) are developed and virus is cleared
(iii) Genotyping: In general, the 5’UTR (untranslated region) of the virus or other highly conserved regions of the virus, such as NS5, or core, are targeted in HCV genotyping. Genotyping techniques include: reverse hybridisation based on differential hybridization of HCV genome segments to DNA probes immobilised on a nylon strip, genotype specific real-time PCR and Sanger sequencing, which has been the most widely available sequencing method since its development in 1977.
Most individuals are infected with a single dominant genotype, but it is possible to have mixed infection with more than one genotype; this has been reported in 2-10% of all HCV positive patients. Commercially available genotyping methods and Sanger sequencing are only capable of reliably identifying the dominant genotype present within a mixed infection sample. Next generation sequencing (NGS) is the current gold standard for the diagnosis of mixed infections. NGS generates millions of sequences in a single run, which allows minority sequences to be detected, improving the sensitivity for the detection of strains that would be undetected by conventional methods. Genotyping studies using NGS show that in general, there is a dominant genotype, with a minor strain contributing to 20% or less of the total number of sequences – these can be missed by conventional assays, including Sanger sequencing (see also later 15.16d).
Decision to test for hepatitis C: As for hepatitis B, there are two approaches to the decision to test for hepatitis infection – either because an individual presents with clinical features that could be consistent with hepatitis (see section 15.8) or because they meet an indication for screening for hepatitis as discussed below.
Individuals may be tested as a result of screening activities when they are asymptomatic (blood donor screening for example), on the basis of risk factors (such as past or current PWID (person who injects drugs), or residence in secure and detained settings), or as part of public awareness raising, such as awareness raising by the Hepatitis C Trust in the South Asian Community and World Hepatitis Day (28th July each year). The CDC (Centre for Communicable Diseases) in the USA has recommended that all adults born during 1945–65 (‘baby-boomers’) receive one-time testing for HCV. The rationale for this was that more than three out of every one hundred baby boomers were infected with HCV, at least five times higher than in any other group of adults, accounting for about 75% of HCV cases in the USA. Risk factor assessments suggest that this group may have been more likely to engage in occasional or ongoing injection drug use during young adulthood, particularly in the 1970s and 1980s. The WHO approach to hepatitis testing in different populations is as detailed in the table in the section on hepatitis above.
NICE guidelines64 recommend hepatitis C screening for the following groups:
For hepatitis C, groups at increased risk include:
In 2015, general practice tested the greatest proportion of individuals for anti-HCV (32.3%), with a further 18.7% tested in other known hospital wards and 16.5% tested in genito-urinary medicine clinics.
Over the last two decades (1996-2017) there has been a more than 8-fold increase in the number of laboratory reports of HCV (positive HCV antibody and/or HCV RNA) in England (see figure below).72
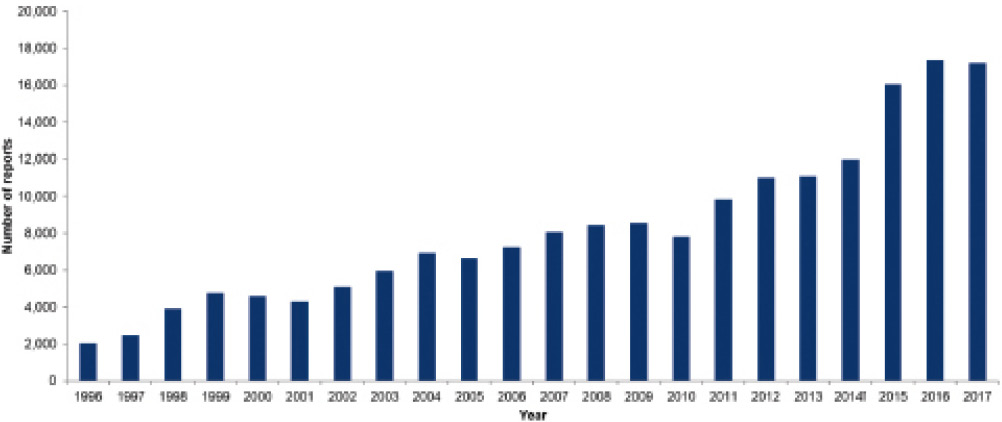
*Laboratory reports include positive test results for hepatitis C antibody and/or hepatitis C RNS: 2017 data are provisional and figures for previous years are subject to change as a result of late reporting and the associated de-duplication procedure. The nature of laboratory reporting and the associated de-duplication procedure is such that re-infections are not captured. In addition, patient identifiable data submitted by NHS laboratories is variable, particularly from sexual health and drug and alcohol services, which limits the ability to de-duplicate. Results for children under 1 year of age are excluded to rule out the likelihood of simply detecting maternal antibody.
† DBS testing from some, but not all private laboratories included from 2014
Data Source : CoSurv/SGSS
Figure 15.6e Laboratory Reports of HCV in England
The number of tests undertaken in sentinel laboratories in England between January 2013 and December 2017 has increased by 20.7%, with an increase of 12.7% in individuals who have not had a previous test. The proportion with a positive test has decreased from 1.9% to 1.6%, consistent with more testing taking place in lower risk populations.
Following the discovery of Australia antigen (AuAg), it became possible to recognise HBV infection in the 1960s using agar gel double diffusion techniques developed by Ouchterlony, but this was a short lived and insensitive diagnostic solution. In the early 1970s radioimmunoassays were developed (EIAs using radioactivity as the detection method); since radioactivity can be detected with high sensitivity these assays had good sensitivity and were adopted for the screening of blood donors and into clinical practice.92 EIA based assays have undergone a series of modifications over subsequent years, mainly based on amplifying methods of detection using different read-out technologies.
The initial screening tests for hepatitis B are now very sensitive; almost every sample that contains the infectious disease agent will test positive. Current commercial third-generation assays have a sensitivity (which compares with HBV-DNA PCR assays) of >95% and in many cases close to 100%. The majority of assays used to detect HBsAg currently in use in the UK are based on chemiluminescence (CLIA/CMIA), and some (such as the Abbott Architect HBsAg Qualitative II assay) provide a quantitative read-out as the amount of chemiluminescence (as measured by relative light units (RLUs)) proportional to the amount of HBsAg in the sample. The Abbott CMIA assay has a reported sensitivity of up to 100% in some studies and a specificity of 99.9% in populations with an assumed zero prevalence of HBV infection. Increasingly HBsAg assays are being designed to detect HBsAg mutations and are able to demonstrate that the assays can detect all known HBV genotypes.
Traditionally the presence of hepatitis B e-antigen was synonymous with a high hepatitis B virus load. Nucleic acid testing for HBV-DNA is now used to quantify HBV viral load, to determine the need for anti-viral therapy in conjunction with ALT levels and degree of liver fibrosis, and to measure the effectiveness of therapeutic agents. HBV DNA is measured in international units (IU)/mL as the recognized international standard or copies/ml by NAT technologies. HBV DNA may be detectable in early infection before HBsAg, and therefore useful in early diagnosis of at-risk individuals before HBsAg appears, depending on the sensitivity of the assay.
The first generation of assays to be developed looked for the presence of a single antibody to a protein antigen (NS4). The sensitivity of these first generation EIAs was low for a high-prevalence population (approximately 80%) and the proportion of positive results that were false positive was as high as 70% for a low-prevalence population (blood donors).93 This led to the development of more sensitive and specific second-generation EIAs (EIAs 2.0) that incorporated additional synthetic or recombinant antigens from the putative core and non-structural regions of the virus (NS3 and NS4); these assays were approved for use by the Food and Drug Administration (FDA) in 1992. These second-generation assays reduced the mean window of seroconversion (time taken from infection to detection of antibody) from 16 weeks to 10 weeks. The sensitivities of second-generation (EIAs 2.0) in a high-prevalence population are approximately 95% (based on HCV RNA detection by PCR).
In 1996, a third-generation EIA (EIA 3.0) was approved by the FDA – this assay added a fourth antigen (NS5) to those in EIAs 2.0, further reducing the window period to around 40 days (WHO: HCV assays, Operational Characteristics 2001). Third-generation EIAs have a sensitivity of around 98%. Reasons for a false-negative result include patients with acute HCV infection (before the HCV antibody has appeared), persons with major immunosuppression (advanced HIV infection or organ transplantation recipients), and persons with chronic renal failure on long-term haemodialysis. The third-generation HCV EIA also has the highest specificity among the EIAs, with a reported specificity greater than 99% in high prevalence populations; false-positive tests can occur with increased gamma globulin production and with autoimmune diseases. False-positive results are also more likely when performing widespread testing in populations that have a very low HCV prevalence. As an example, the Abbott Architect anti-HCV assay is CMIA based and detects antibodies to antigens from portions of the core coding region of HCV, NS3 and NS4 regions of the virus. Company data reports an overall sensitivity of 99.10% (95% confidence interval 96.77-99.89%), and specificity depending on the population tested from 99.20% and 99.70%. Testing strategies have evolved so that all samples testing positive are tested using an alternative assay; either by using a second/alternative antibody assay, and/or looking for evidence of the virus itself by using molecular assays to detect viral RNA, or ELISA based assays to detect HCV antigen.
Donor selection criteria aim to ensure that the population eligible to donate blood are at low risk for viral infections which have potential to be transmitted via transfusion. Data confirm this to be an effective strategy; the UK prevalence of HCV infection is 0.67%, but the prevalence among first time blood donors meeting donor selection criteria is 0.03%. Testing strategies employed to assess the suitability of donated blood for transfusion are therefore used in the context of very low chance of infection, but given that it is essential that all infections are detected, assays with high sensitivity are used.
Hepatitis B: HBsAg testing was initially implemented by the blood services in the 1970s, with NAT (HBV DNA) based assays introduced in 2009. Initial serological assays were less sensitive than those used now. The UK specification for minimum sensitivity for HBsAg screening is 0.2IU/mL76 – this is the assay that will detect HBsAg at that concentration in a blood sample. A UK standard of this concentration is available from the National Institute for Biological Standards and Control (NIBSC), which must be included in these assays as a control, in addition to manufacturer’s controls. Each batch of assays must be demonstrated to conform to these requirements prior to use. HBsAg assays used by the blood services estimate a sensitivity of 100% with specificity of 99.99%.
As discussed above, NAT assays are employed to identify the presence of HBV-DNA within a sample which may identify cases of early infection in which HBsAg is not yet positive. There is no specific UK requirement for the minimum sensitivity of HBV NAT; however, a standard is available and all assays must be appropriately controlled. Samples from individual donations are pooled for initial testing. If a pool is reactive the samples that make up the pool are tested individually to identify the reactive donation(s).
Hepatitis C: Anti-HCV serology was introduced by the blood services in the early 1990s, with NAT for HCV RNA introduced in 1999. The UK requirement for the minimum level of sensitivity for the performance of anti-HCV screening is that a positive result should be obtained with the UK anti-HCV working standard available from the NIBSC. Assays used by the blood services currently estimate a specificity of 99.73% and sensitivity of 100%.
The UK requirement for the minimum level of sensitivity for the performance of HCV NAT is 5,000 IU/mL in an individual donation.76 An HCV RNA international standard is available from the NIBSC. Similar to HBV NAT, donor samples are initially pooled for testing, and if a pool is reactive, the individual samples will be analysed to identify the reactive donation(s).
There is a very wide spectrum of signs and symptoms experienced by patients during the first phase of infection with both hepatitis B and hepatitis C. A large proportion of patients do not have any symptoms or signs at all. This depends on the virus and the age of the person. In children under the age of 5 more than 90% will be free from symptoms and signs of disease. They and their parents will be completely unaware of the infection. In older children and adults symptoms and signs occur in 30% of those with hepatitis B virus infection and 20-35% of those with hepatitis C virus infection.
Where symptoms occur, there are mild constitutional symptoms such as nausea, loss of appetite, fatigue and vague abdominal pain. Some patients suffer with a skin rash, muscle aches or joint pains. There may be a dull pain in the right upper quadrant over the abdomen. Symptomatic patients generally complain of jaundice; yellowing of the eyes and dark urine.
The signs detected during the acute phase of infection may include jaundice, tenderness over the liver and a patchy red rash over the trunk or the whole body. Symptoms and signs usually occur 2.5–8 weeks after exposure in patients infected with hepatitis C. In patients infected with hepatitis B the symptoms may occur between 6 weeks and 6 months after exposure to the virus. Symptoms and signs usually resolve spontaneously after 1–2 weeks. Rarely, symptoms will persist for 2 months.
As the majority of patients do not have any symptoms or signs during the acute phase of infection, presentation to clinicians only occurs if the patient happens to have a blood test for liver biochemistry (known as liver function tests (LFTs)) that would indicate the presence of liver inflammation. If the patient was thought to be at risk of a hepatitis virus infection they would be tested using tests specific for the hepatitis viruses. Patients who become symptomatic after infection would present with the symptoms described above. In the majority of cases patients’ symptoms develop over a few days and they are seen in primary care by their General Practitioner. Occasionally the symptoms develop rapidly or are severe leading to attendance at an accident and emergency department.
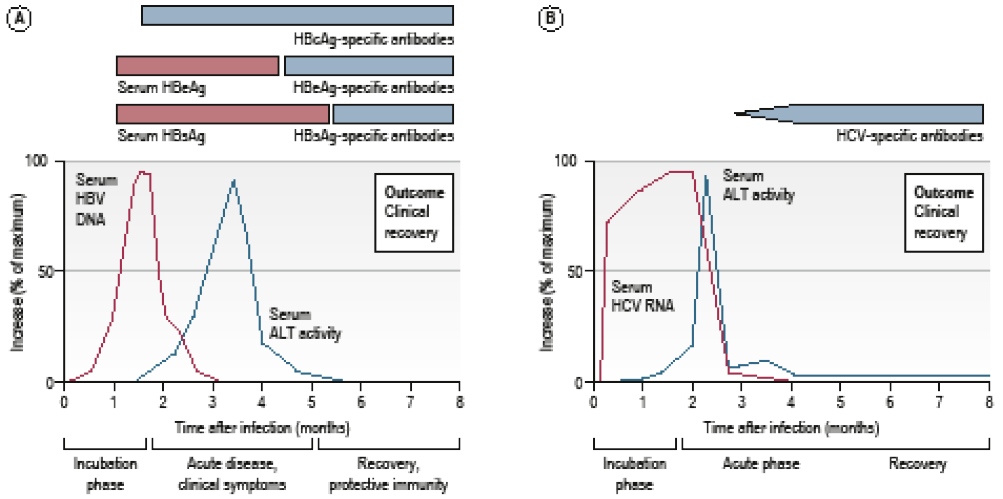
Figure 15.8 (a,b) Course of self-limiting acute infection with (a) HBV (b) HCV (from Manson’s Tropical Diseases, 23rd edition, adapted from Rehermann and Nascimbeni. Nature Reviews Immunology 2005; 5:215 – 29.)
The term ‘acute’ is used to describe a short duration of illness, in contrast to the term ‘chronic’ which denotes an illness of long duration or one which persists indefinitely. By consensus an acute hepatitis infection has a maximum duration of 6 months, so any infection which persists longer than this is considered as chronic infection. Nevertheless, all chronic infections have an acute phase which lasts up to 6 months. ‘Acute’ and ‘chronic’ do not signify anything about the severity of the infection, nor do these terms indicate whether the infection causes symptoms.
It is not entirely clear why some people spontaneously clear viral hepatitis infections whilst others progress to chronic infection. We know that the age at which the infection is acquired is very important, particularly with hepatitis B virus infection. Gender is another important factor: females clear spontaneously more frequently than males. Genetic variants also contribute to the likelihood of clearing the infection. People who are immunocompromised due to genetic defects (such as agammaglobulinaemia), treatment for cancer, immunosuppressive drugs or infection with HIV are more likely to develop chronic infection.
Acute hepatitis may be associated with symptoms and signs as described in section 15.8. These range in severity from a minor ‘flu-like’ illness accompanied by mild jaundice through to a severe illness characterised by abdominal pain, deep jaundice, joint and muscle pains and, in a very few cases, signs of liver failure, such as confusion and coma. In patients infected with the hepatitis B virus, the occurrence of signs and symptoms in the acute phase is associated with a much lower chance of the infection becoming chronic. Signs and symptoms are much more likely to occur in patients who are adult and the risk of chronic infection in this situation is less than 5%. Similarly, the risk of chronic infection is substantially reduced in patients infected with hepatitis C virus where the acute phase is symptomatic.
There is no evidence to suggest that symptoms in the acute phase have any impact on disease severity if the infection progresses into the chronic phase.
Physical signs and symptoms associated with the acute infection are described in section 15.9 above. The vast majority of patients with chronic HBV or HCV infection have no symptoms at all. Measurement of quality of life using questionnaires completed by patients indicate a reduction in mental well-being and physical functioning amongst patients chronically infected with HCV.94,95 However, some of the studies showing a reduction in quality of life may be considered unreliable because the questionnaires were conducted after the patients had received their diagnosis. The diminished quality of life may, therefore, be a result of knowing the diagnosis and prognosis rather than a direct impact of the infection. It is widely accepted that some patients with chronic HCV infection suffer from neurocognitive symptoms which may include fatigue, anxiety, depression, problems with cognition known as “brain fog”, attention deficit and memory impairment. These symptoms are associated with low level inflammation in the brain and with functional changes which can be identified using specialised MRI scans. Many of these neurocognitive problems can persist even after successful treatment of the infection.94
Pain and tenderness over the liver, fevers, dyspepsia and irritable bowel symptoms are not recognised features of chronic viral hepatitis infection.
A rare complication of HCV infection is a condition called essential cryoglobulinemia which is associated with a skin rash and peripheral nerve damage, and loss of sensation in the fingers. Cryoglobulinemia resolves once the infection is cured, but damage to the nerves may not improve.
Chronic viral hepatitis is not known to cause dizziness, high blood pressure, sweating, weight gain, weight loss, anal bleeding or urges to eat sugary foods. These symptoms can also be very common in people who are not infected.
When patients develop advanced liver disease with cirrhosis, loss of liver function and increasing pressure in the abdominal venous system cause a variety of symptoms and signs. These include abdominal swelling due to fluid collection (known as ascites), jaundice, confusion and coma (known as encephalopathy), vomiting blood or passage of altered blood in the stool due to bleeding veins in the oesophagus, fatigue, breathlessness and susceptibility to bruising due to loss of clotting factors. These symptoms and signs occur at the stage of disease known as decompensated cirrhosis and are associated with limited life expectancy unless the patient receives a liver transplant.
Alcohol use is fairly common amongst patients with chronic viral hepatitis infections. It can be difficult to distinguish whether alcohol or the virus are responsible for the liver inflammation, as represented by abnormal liver function tests, or the liver fibrosis. Symptoms and signs are not helpful to distinguish the dominant cause of liver damage. Skilled pathologists may be able to distinguish the most important cause of damage on the liver biopsy, but in the majority of cases viral infection and alcohol excess work together to exacerbate liver damage.
The natural history of HBV depends to a great extent on the age of exposure. Exposure to HBV during birth or early childhood usually occurs without any symptoms and more than 90% of those affected develop chronic infection. This mode of transmission is common in populations where there is high frequency of HBV infection, for example sub-Saharan Africa. The lifetime risk of cirrhosis in people infected in infancy is estimated at 15-40%. In early adulthood the virus is usually present at high level, but does not cause liver inflammation or damage; this is termed the immune tolerant phase. It is of variable duration and can last decades. Over time patients may develop liver injury as the immune system interacts with the virus, termed the immune active phase. Liver injury may persist and cirrhosis can develop.
Following exposure to HCV most people will not experience any symptoms or signs and are, therefore, unaware that they have contracted the virus. Less than 20% of patients experience the typical symptoms of acute hepatitis, such as malaise, fatigue and jaundice. The virus then persists in the liver and can silently cause liver inflammation and scarring. Over time, the liver scarring can become more severe leading to cirrhosis, when the liver structure forms into nodules surrounded by scar tissue.
Liver cirrhosis is the end result of long-standing liver disease of any cause. Cirrhosis can be defined as advanced liver scarring. The normal microscopic structure of the liver is no longer present and instead nodules of liver cells are surrounded by bands of fibrous scar tissue (hence fibrosis). The images below show liver samples at different stages of scarring. In a normal liver fine strands of fibrous tissue (staining black) give a support structure to the liver cells and blood vessels. The thicker bands of tissue initially start next to the blood vessels and then join to form thick bands surrounding liver cells. The liver may continue to function well in the presence of cirrhosis (this is termed compensated or early cirrhosis and can be scored for severity according to Child-Pugh criteria). However, over time the cirrhosis severity can progress. The common potential complications of cirrhosis are due to loss of function of liver cells and an increase in the pressure within blood vessels taking blood from the intestine to the liver, termed portal hypertension. The four common complications of cirrhosis are ascites (abdominal fluid retention), encephalopathy (fluctuating confusion due to reduced removal of toxins from the blood), varices (varicose veins that form in the intestine) and liver cell cancer. The development of ascites, jaundice or encephalopathy is termed decompensated cirrhosis.
In addition, it is important to note that patients may have disordered blood due to advanced liver disease per se. This is in part a consequence of reduced production of proteins (clotting factors) and low levels of platelets (the component of blood involved in clotting).
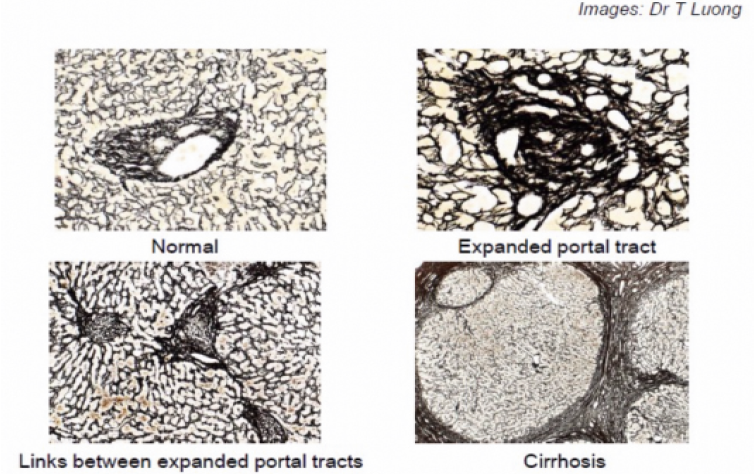
Figure 15.11a shows typical images of a liver biopsy viewed under a microscope (i) (top left) normal liver; (ii) (top right) early changes in the liver associated with inflammation; (iii) (bottom left) normal liver, but with expanding areas of fibrosis/scarring that begin to connect; (iv) (bottom right) cirrhosis – islands of liver surrounded by thick bands of fibrous material
Cryoglobulins are abnormal immune proteins. Both HCV and HBV can lead to production of cryoglobulins. When present in the blood they can cause a variety of problems, such as skin rash, joint pains or kidney damage.
Chronic infection with hepatitis C is associated with a small increased risk of lymphoma.
HBV
The 5-year cumulative incidence of cirrhosis range is 8%-20% in untreated patients with chronic HBV and the 5-year cumulative risk of liver failure (hepatic decompensation) in those with cirrhosis is 20%.96 A range of factors have been associated with more rapid progression of liver disease, including greater liver inflammation, older age, high alcohol intake, co-infections (particularly HDV and HIV), diabetes and obesity. In general, the risk of liver cancer developing is higher with more advanced liver disease and is markedly reduced by effective antiviral therapy. For individuals without advanced liver disease annual risk of hepatocellular carcinoma (HCC) has been estimated at 0.01-1.4% a year, with reductions of up to 80% from effective treatment. For individuals with HBV and cirrhosis, rates are, on average, higher (0.9%-5.4%) with more modest reduction from treatment (approx 30%).97 US Guidelines recommend surveillance for HCC if annual risk of HCC > 0.2%.98 In effect this means most of those infected with HBV are recommended to have six monthly testing (usually involving an ultrasound of the liver and an AFP (alpha-feto protein) blood test).
HCV
Estimates of the rate of progression from infection to cirrhosis vary widely, but have been estimated at 1-2%/year,99 with approximately 20-30% with cirrhosis after 20 years (but estimates range from 2-40% in different studies) and 40% at 30 years.100 Several factors, similar to those for HBV outlined above (with the exception of HDV) are associated with more rapid progression.
The risk of developing liver cancer has been estimated at 2-8% a year in those with cirrhosis and is significantly lower (<1% per year) in those with less advanced disease. Successful treatment for HCV can considerably reduce (by approximately 70%), but not eliminate, the risk of cancer. US guidelines have a higher threshold compared to HBV for recommending screening for liver cancer (1.5% per year), which means screening is recommended for those with advanced disease, even after successful treatment.98
There are many studies that have investigated the effect of HBV and HCV on disease and life expectancy. It is difficult to make an estimate of the prognosis for an individual as progression of disease depends on a range of factors, including the virus and the genetics of the person infected, but more importantly, whether other factors are present that might influence progress. These include other medical conditions, illicit drug use or the levels of alcohol intake, as well as access to effective therapy and the presence of other conditions that might limit life expectancy. In addition, individuals infected at different times of life in different parts of the world will have different outcomes from their infection. In general, death certificates tend to underreport deaths due to viral hepatitis.101
In 2013, the estimated 1.4 million deaths related to viral hepatitis globally equated to 42 million years of life lost.2 In a large study of individuals living with HBV in China, life expectancy was reduced, on average, to 71.8 years in men (compared to 76.2 years in those not infected).102 A smaller reduction was observed in women (82.0 to 80.1 years).103
A study in New York found that of individuals infected with HBV, 55% were likely to die prematurely (aged <65)104 as were those infected with HCV, who were not only more likely to die of liver cancer or advanced liver disease, but also drug-related causes.105An Australian study found that HCV reduced life expectancy by an average of approximately 6 years even when drug-related deaths were excluded. 106
A Dutch study107 of haemophiliacs (between 1992 and 2001) found that those without hepatitis or HIV co-infection had a similar life expectancy to the general population (average 74 years compared to 76);108 however, those infected with HCV had mortality rates 16 times higher.
For all causes of liver cirrhosis, treatment of the underlying cause of cirrhosis and avoidance of any co-factors, such as alcohol consumption, is important. Treatment for specific complications is outlined below. Ascites and encephalopathy are features of decompensated cirrhosis and develop in approximately 20% of patients with cirrhosis within 5 years after diagnosis. Varices and HCC can develop in patients with compensated or decompensated cirrhosis.
Ascites is fluid which collects within the abdomen causing distension and discomfort. Liver cirrhosis is the commonest cause of ascites, but it can also be caused by heart failure, kidney failure or cancer within the abdomen.
The approach to ascites treatment is stepwise. First, general advice includes restriction of dietary salt intake and commencement of diuretic tablets, usually spironolactone alone or in combination with furosemide. Most patients will respond to diuretics with resolution of ascites. It is also important to ensure dietary advice has been given, as loss of weight and muscle loss are common in patients with ascites.
Some patients do not respond, or have side effects from diuretics, such as low blood sodium or impaired kidney function. These patients are described as having “refractory” or “resistant” ascites. The question of whether a patient is suitable for liver transplantation should be asked for all patients who develop refractory ascites. If the patient is a suitable candidate for a liver transplant they should be referred to a liver transplant centre.
Many patients may have reasons why liver transplantation is not feasible, often due to other health conditions or frailty. For those patients the treatment options are as follows:
A future option may be an implanted pump which sits within the abdomen and pumps ascites fluid into the bladder via a tube. The ascites is then passed out in the urine. This device has been evaluated in clinical trials. In November 2018, NICE provided guidance on the use of these pumps as follows: “Current evidence on the safety of subcutaneous automated low-flow pump implantation for refractory ascites shows there are serious but well-recognised safety concerns, including device failure and acute kidney injury. Evidence on efficacy is limited in quantity. Therefore, this procedure should only be used with special arrangements for clinical governance, consent, and audit or research.”
Varices are large venous channels that develop in the intestine as a result of portal hypertension (back pressure in blood vessels draining into a cirrhotic liver). The most common sites are the oesophagus, stomach and rectum, but they can occur anywhere in the intestine.
The main complication of varices is bleeding, which can be life-threatening. Currently, it is recommended that patients diagnosed with cirrhosis have an upper GI endoscopy to look for the presence of gastro-oesophageal varices.
If none are present the endoscopy can be repeated in 2-3 years. If small oesophageal varices are present and the patient has no history of bleeding, no intervention is needed, but the endoscopy should be repeated in 1 year to reassess the size of varices.
If medium or large oesophageal varices are present, and the patient has no history of bleeding, the first line treatment is a beta blocker (commonly propranolol, recently carvedilol) that reduces the variceal pressure and reduces the risk of bleeding.
If the patient has medium or large oesophageal varices and is unable to take beta blockers or has side effects, then they should be offered the option of variceal banding. This procedure is done during an endoscopy. A small rubber band is applied to each varix. This stops the flow through the varix which then thromboses and scars. The banding procedure usually needs to be repeated to ensure the varices are treated effectively.
Patients with gastric varices who have never bled may be offered beta blockers or a variceal glue injection if they are considered at high risk of bleeding.
Variceal bleeding is a medical emergency and the patient should be treated in hospital. They may require transfusion of blood and/or clotting factors in an intensive care unit for organ support and monitoring. An urgent endoscopy must be done. The current first line treatment to stop bleeding is endoscopic variceal banding for oesophageal varices and glue injection for gastric varices. If the endoscopic treatment does not stop the bleeding then a TIPS shunt is the next step.
Previously, surgery to create a shunt between the portal vein and a systemic vein or oesophageal transection were treatments used in refractory variceal bleeding. However, these operations had a high risk of complications or mortality. They are very rarely done nowadays due to the development of more effective endoscopic therapies and TIPS shunts.
This is a type of fluctuating confusion due to impaired removal of toxins from the blood by the liver. It can be present in patients with cirrhosis who have poor liver function or if the body has developed spontaneous shunts to bypass the liver. HE can range from mild symptoms, such as impaired concentration or forgetfulness, to drowsiness and coma.
It can be precipitated by other problems, commonly constipation, dehydration, infection, gastrointestinal bleeding or the use of sedative medications. It is important to diagnose and treat the underlying cause. A patient with more severe encephalopathy would need to be admitted to hospital and if a coma develops would be treated on intensive care.
It is important to diagnose and treat the underlying cause of HE as this will help to resolve the symptoms.
Some patients have more persistent symptoms or recurrent episodes of HE. These patients are advised to take regular lactulose which prevents constipation. If there have been hospital admissions due to HE, a non-absorbed antibiotic, rifaximin has been shown to reduce HE symptoms and is approved by NICE. In the past neomycin was used and protein restriction recommended, but these are no longer considered beneficial.
If patients with HE have good liver function then an abdominal scan to look for the presence of spontaneous portosystemic shunts should be done. A multicentre study published in 2013 showed an improvement in symptoms of HE following shunt blockage in patients with cirrhosis and HE with good liver function.
HE being refractory to medical treatment is an indication for liver transplantation, and if patients are suitable candidates they should be referred to a liver transplant centre. All patients with HE should be advised to contact the Driver and Vehicle Licensing Agency (DVLA), as they should not drive.
Liver transplantation is now a well-established treatment for decompensated end-stage liver disease. The first liver transplant in the UK was carried out in 1968. The number of liver transplants started to increase in the late 1980s due to improvements in medical and surgical care. In 2018 more than 1,000 liver transplants were performed in the UK. It is expected that one year survival after a liver transplant is >91% and 5 year survival >80%. In the 1990s, hepatitis B transplant patients had worse survival when compared to those with other liver diseases, due to recurrent HBV infection. Since the development of effective drugs for HBV, such as tenofovir and entecavir, the post-transplant survival for patients with hepatitis B is better than average.
Similarly, prior to 2014, patients receiving a liver transplant for hepatitis C had lower survival due to recurrence of HCV after the transplant. Approximately 10% of patients experienced rapid progression to cirrhosis within 1-2 years, termed fibrosing cholestatic hepatitis. In other patients, although fibrosis progression was slower, it was still faster than in the pre-transplant population, with an average time to progress to cirrhosis of 5-8 years after transplantation. Interferon-based regimes were less effective and less well tolerated in patients after transplantation. Since the development of direct acting antiviral treatment for HCV, recurrent HCV infection after the transplant is now very rare, occurring in less than 2% of patients.
Consideration of liver transplantation should happen for any patient with cirrhosis who has an episode of decompensation, such as ascites or hepatic encephalopathy. Liver transplantation is major surgery with a need for lifelong monitoring and patients need to undergo assessment of their fitness for the surgery. Patients with decompensated cirrhosis should be referred to a liver transplant centre unless they are clearly not fit to undergo major surgery.
Patients with cirrhosis are at risk of developing primary liver cancer, termed hepatocellular carcinoma (HCC). The risk for patients with cirrhosis due to chronic viral hepatitis B or C is approximately 2-8% per year. Antiviral treatment is likely to lower this risk by approximately 50-70%. Patients with HBV or HCV who do not have cirrhosis have an annual risk of approximately 0.2% for developing HCC.
Guidelines recommend offering surveillance tests to look for early HCC in patients with cirrhosis. The usual tests are liver ultrasound (US) performed every 6 months. Serum alpha-feto protein (AFP) can also be measured, but is not abnormal in all patients with HCC and is not recommended in all guidelines (see Q15.24).
Decisions about treatment for HCC need to consider the stage of HCC, severity of liver cirrhosis and the patient’s general fitness relating to liver and other health conditions.
The Barcelona Clinic Liver Cancer (BCLC) staging system incorporates these factors and is the most commonly used algorithm. The diagram below indicates the BCLC algorithm and the expected survival associated with each treatment option. The estimates of mortality include mortality due to progression of cancer or progression of cirrhosis. Liver resection, liver transplantation and thermal ablation are generally considered to be curative therapies. Systemic therapies and transarterial chemoembolisation (TACE) are generally considered as palliative therapies.
In common with most Western countries, most patients with HCC are diagnosed at an advanced stage and can only receive palliative therapies. Approximately 15-20% of UK patients are diagnosed with early stage HCC and can be considered for curative therapies. The main reason for late diagnosis of HCC is that many people with cirrhosis are not aware they have this condition and are therefore not offered surveillance. In addition, surveillance tests lack accuracy and are not consistently offered or performed for patients with cirrhosis across the UK.
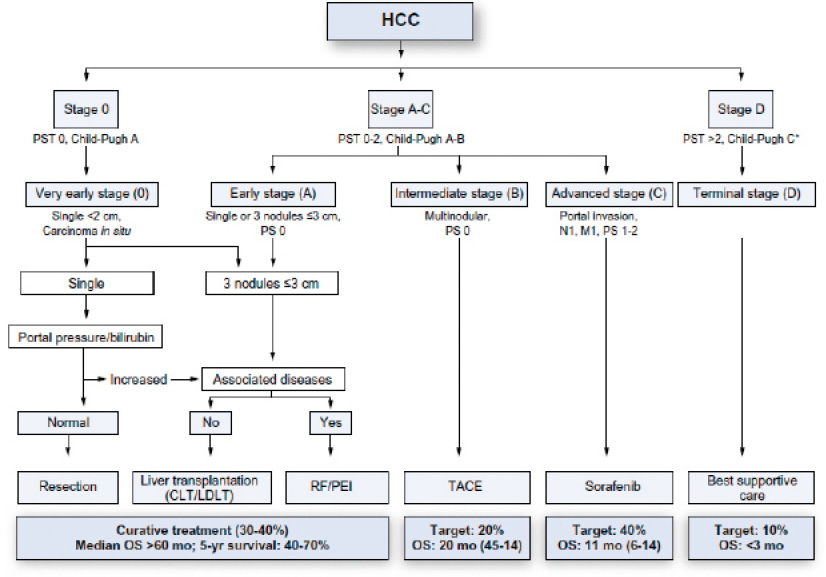
Figure 15.11b
This treatment is surgery to remove the part of the liver that contains the HCC. This treatment is usually offered to patients who do not have liver cirrhosis, or those patients with cirrhosis who have no evidence of portal hypertension and have good liver function. The risk of recurrence of HCC or development of a new HCC in the cirrhotic liver is approximately 50-70% at 5 years.
This treatment is performed using heat or microwave energy to destroy cancer cells. Under general anaesthetic a probe is inserted into the tumour and heat or microwaves applied. This treatment is best for a signal HCC that is smaller than 3 cm in size. The risk of recurrence or a new tumour is also 50-70% at 5 years. Thermal ablation for HCC has been described in the literature since 1995.
Approximately 20-25% of UK patients receiving liver transplant have HCC. HCC was considered an indication for liver transplant from the beginning of liver transplantation in the 1960s. However, it soon became apparent that patients with advanced stage HCC were at high risk of HCC recurrence after the transplant, which has a high mortality. Criteria based on size and number of HCC, to minimise this risk, were introduced based on research published by Mazzaferro and colleagues in 1995, termed the Milan criteria. The UK criteria have expanded modestly since then, such that patients with a single HCC <5cm, or up to 5 HCC <3cm, or a single HCC 5-7 cm that remains stable in size over 6 months, can be considered for transplant. At present there is a pilot programme evaluating the benefit of liver transplantation in patients with HCC that initially exceed these criteria but have a response to other cancer treatments such that the tumours become smaller and meet those criteria.
As with decompensated cirrhosis, patients being considered for liver transplantation for HCC need to be fit to undergo this major surgery, taking into account any other medical conditions.
HCC are vascular cancers that receive blood supply mainly from the hepatic artery, whereas the rest of the liver depends primarily on blood supply from the portal vein (draining the intestines). Therefore, blocking the hepatic artery supplying an HCC would starve the cancer of oxygen and nutrients (leading to cancer cell death), but have less adverse effect on the background liver. Sometimes, chemotherapy drugs or beads coated with chemotherapy drugs can be injected at the same time. TACE has been described in the literature since 1982. It is the treatment used most frequently for HCC in the UK.
Drug therapies licensed and available to treat HCC are in the group of drugs named tyrosine kinase inhibitors (including sorafenib, lenvatinib and regorafenib). These drugs target the pathways that promote cancer cell growth and the new blood vessels that grow to supply liver cancers.
Sorafenib was the first drug to demonstrate improved survival in patients with HCC in research published in 2008. The drug was made available in England in 2010 through the Cancer Drugs Fund. It was approved by NICE for the treatment of HCC in 2017.
Lenvatinib was approved by NICE for HCC in December 2018 as first line treatment. Regorafenib was approved in January 2019 as second line treatment for patients who have progressed on sorafenib.
New treatments for HCC are being evaluated in clinical trials. Immunotherapy drugs appear promising, with good results reported in some patients. As yet no immunotherapies have been approved in the UK for HCC and patients can only receive these treatments through participation in clinical trials.
There are two main techniques for applying radiotherapy to HCC, trans arterial radioembolisation (hepatic artery injection of beads coated with radioactive isotopes) or stereotactic external beam radiotherapy, where radiation is concentrated from an external source into the cancer. At present neither of these are approved by NICE for treatment of HCC.
Palliative care is defined by the WHO as “an approach that improves the quality of life of patients and their families facing the problem associated with life-threatening illness, through the prevention and relief of suffering by means of early identification and impeccable assessment and treatment of pain and other problems”.
Traditionally, the diagnosis of incurable cancer has been a trigger to prompt referral to specialist palliative care, which may be in hospital or hospice. Patients with advanced decompensated cirrhosis, who may have a similar prognosis, have in the past been referred very late for palliative care, often in the last days of life.
In the last 5 years there has been increasing recognition that patients with non-cancer life-limiting conditions, such as decompensated cirrhosis, could benefit from specialist palliative care (SPC). Tools have been developed to help identify patients with cirrhosis who could benefit, such as the Bristol Score or SPICT (the Supportive and Palliative Care Indicators Tool).109 In addition to addressing symptom control at the end of life, referral to palliative care within the last year of life can also address holistic needs and advanced care planning.
There is variability throughout the UK in referral to SPC and access to SPC services for patients with chronic liver disease.
In general, individuals who have been living with HBV or HCV for longer periods of time are more likely to have developed progressive scarring (fibrosis, even cirrhosis) of the liver. In the setting of fibrosis, the likelihood of developing end-stage liver disease or liver cancer is higher. The risk of liver cancer and progression to end-stage liver disease is reduced by effective treatment for both HBV and HCV with the potential for reversing fibrosis once it has developed. However, with advanced fibrosis, improvements following successful treatment may be small. In general, treatment is best initiated before fibrosis develops.
There are differences in the current approach to treatment for HBV and HCV. Not all HBV patients are recommended to have treatment, given that some patients are unlikely to progress to fibrosis, and the challenge of taking long-term treatment that carries potential toxicity, with the potential benefit of therapy, must be weighed up. Current international guidelines recommend antiviral treatment for patients with cirrhosis and patients without cirrhosis who have elevated liver enzymes and elevated blood HBV DNA above certain thresholds. Patients who do not meet these criteria should be monitored as their condition may evolve to meet treatment criteria.96 Over time this approach has been consistent, but where previously decisions were based more on biopsies of the liver, there is now greater emphasis on blood tests and other, less invasive, means of assessing the liver.
For HCV the current treatment regimes are finite, highly effective at curing the virus and have few side effects; therefore all patients with chronic HCV who are well enough to receive treatment should be offered it. The benefit for patients without cirrhosis is the prevention of progressive liver disease. Even in patients with advanced cirrhosis, improvement can be seen in their liver function. Shortly after the introduction of DAA therapy there was some controversy, after some studies suggested an increase in HCC risk after treatment in those with the most advanced disease. However, following further research, this is now not thought to occur and it is likely that treatment would reduce the long-term risk of HCC.110
Prior to the current developments in HCV therapy, not all patients would necessarily have been recommended for therapy. Prior to 2014, treatment regimes were based on interferon, which was less effective and associated with more side effects, especially in patients with cirrhosis (see 15.13). Therefore, it was common to monitor patients with mild liver disease due to HCV and offer antiviral treatment to patients with evidence of fibrosis development. Patients with compensated cirrhosis could be given treatment. but with a potential side effect of precipitating decompensation.
There is clear evidence that repeated exposure to different HCV viral strains can lead to multiple infections in individuals.111 Although it has been suggested that multiple infections could lead to more rapid disease progression, there is little clinical evidence to support this. However, it is reasonable to think that multiple infection is more likely to lead to infection with specific viral genotypes associated with poorer outcomes for treatment (notably genotypes 1 and 4 in the era of interferon therapy, and genotype 3 in the DAA era).
There is evidence that individuals infected with multiple genotypes of HCV do not have higher viral loads than those infected with single genotypes.112
Q15.12(d) is addressed in Q15.16(c)-(d).
Q15.12(e) is addressed in Q15.16(a)-(b).
Although hepatitis B virus (HBV) was first discovered in 1965, the first treatment for hepatitis B was not approved until 1992. That first treatment, interferon, still has a niche role in therapy, but has been replaced in mainstream treatment by oral medication that targets the replication of the virus. The most commonly used are nucleoside/nucleotide therapies, in particular, tenofovir disproxil fumarate (TDF) and entecavir. No drugs for hepatitis B are curative and this is an area of active research (see later Q15.25). This means most treatments are continued life-long once started. All the drugs discussed in the following sections have detailed summaries of product characteristics (SPCs) available with detailed descriptions of action, side effects and interactions, only the key features of which are described here.
Interferons were used experimentally from 1981 and were used in routine clinical practice when a genetically engineered (also called recombinant) interferon product became approved for treatment of HBV in 1992. Interferons had to be administered three times a week by injection under the skin. Attaching a polyethylene glycol (PEG) molecule to the interferon created a pegylated interferon which had a longer lasting effect and therefore could be administered once a week. The peglyated version (PEG-IFNa) was approved for use in 2002. Unlike in the treatment of hepatitis C, its use in hepatitis B was more limited, although treatment with PEG-IFN has been recommended by NICE since 2013 for some patients.
In order to understand the rationale and goals of treatment, it is important to recognise the four phases of chronic HBV infection:
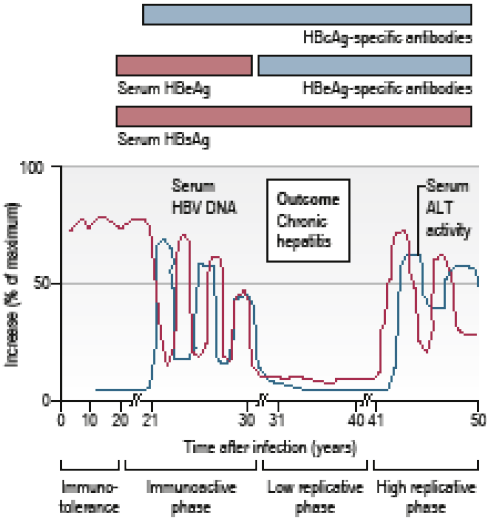
Figure 15.13a Illustrative phases of chronic HBV infection following infection at birth (from Manson’s Tropical Diseases, 23rd edition, adapted from Rehermann and Nascimbeni. Nature Reviews Immunology 2005;5:215 – 29.)
Pegylated interferon is used to move patients from either the immune reactive HBeAg-positive phase or the HBeAg-negative chronic hepatitis B phase to the inactive HBV carrier state. This may result in long-term control of viral replication and prevention of further liver fibrosis. This treatment goal is achieved in around 30% of patients who are treated in the immune reactive HBeAg-positive phase and in around 20% of patients who are treated in the HBeAg-negative chronic hepatitis B phase. The patients who are most likely to respond well to treatment are female, younger age, HBeAg-positive and those infected with favourable strains of the virus (eg HBV genotype A).
The advantage of interferon treatment is that in contrast to oral nucleoside/nucleotide therapies (see below), treatment is finite, typically recommended for 48 weeks. In the small numbers of patients who benefit, this treatment may mean they do not require prolonged therapy with oral treatments. However, the length of the course and the significant side effects associated with prolonged courses of treatment (see Table 15.13a and elsewhere in this letter) mean its use is rare.
Direct antivirals are drugs that block virus replication within the cell. HBV creates its own little chromosome in the nucleus of the cell, so stopping the virus from replicating does not result in a cure. Nevertheless suppressing the virus results in complete resolution of the inflammation in the liver, cessation of liver fibrosis and subsequently reversal of the fibrosis and a marked reduction in the risk of developing liver cancer. However, direct antiviral drugs have to be taken indefinitely. With very few exceptions it would be anticipated that all patients who start antiviral drugs would remain on treatment for the rest of their lives.
Lamivudine (also known as 3TC, trade name Epivir) was originally developed as a treatment for HIV, but also has activity against hepatitis B. It is an oral medication (a nucleoside analogue of cytidine) which inhibits reverse transcriptase. It was approved by the FDA in 1998 and became part of NICE recommended therapy for chronic hepatitis B. The key advantages of lamivudine are its simplicity of dosing for patients and its very good side-effect profile, with adverse events rarely requiring changes in therapy. The main disadvantage was that the emergence of a resistant virus, which leads to treatment failure, was common, usually affecting 25% of patients by year 1 and the majority of patients within 2-3 years. This vulnerability of the drug to resistance has led it to fall out of favour when compared to newer nucleoside/nucleotide therapies with lower rates of resistance. It remains a common part of therapy for many HIV-infected individuals who have co-infection with hepatitis, as it provides activity against both viruses.
Adefovir (trade name Hepsera) is a daily oral medication (a nucleotide analogue of adenine) which targets the reverse transcriptase of hepatitis B. It was approved by the FDA for treatment of hepatitis B in 2002 and its use was recommended in a NICE technical assessment in 2006. ADV provided a useful addition to LAM in the treatment of chronic hepatitis B. It was effective as a single treatment agent, but was commonly used as an addition to LAM when resistance began to emerge. In some cases with high levels of viral replication, a combination of LAM/ADV was used. The key drawback of adefovir is its toxicity, particularly to the kidneys. Mild to moderate increases in serum creatinine (a marker of kidney function) were common and severe cases of kidney toxicity also reported. Patients commonly develop tubular damages causing leakage of small quantities of protein in the urine. This toxicity meant the drug was used at relatively low doses (5 to 10mg daily), but despite this, toxicity has limited its use and it is now rarely, if ever, prescribed for hepatitis B.
Entecavir (trade name Baraclude) is an oral medication taken once a day. It is a nucleoside analogue of guanosine which inhibits the reverse transcriptase of HBV. It was first approved by the FDA for treatment of chronic HBV in 2005 and recommended in a NICE technical assessment in 2008. It remains a first line option for treatment in 2019 and is now off patent in the UK.
It found early use in the treatment of patients failing therapy with LAM and/or adefovir, where it was able to suppress resistant virus at a dose of 1mg/day. For patients starting treatment for the first time, a lower dose of 0.5mg is usually recommended. Side effects from entecavir are relatively uncommon and can include metabolic disturbance and fat accumulation in the liver (steatosis), as well as milder side effects of headache, fatigue and dizziness.
Although entecavir has some activity against HIV, its efficacy is not high and it is generally not used for its HIV activity in patients with both HIV and HBV (in contrast to tenofovir based drugs (TDF/TAF)).
Telbivudine (trade name Sebivo) is an oral medication. It is a nucleoside analogue of thymidine and inhibits the reverse transcriptase of HBV. It was first approved by the FDA in 2006 and subject to a NICE assessment in 2008. Although telbivudine offers advantages over lamivudine with greater efficacy and less emergence of viral resistance, it is generally not widely prescribed due to the availability of better treatment options (such as entecavir or tenofovir). Side effects include headache, cough, diarrhoea, nausea, abdominal pain, rash and fatigue.
TDF (trade name Viread) remains one of the most widely used medications in the world for the treatment of HBV. It is a pro-drug, converted in the body to tenofovir, a nucleotide analogue of adenosine that inhibits the replication of HBV. It is dosed once a day and side effects are relatively uncommon. Most importantly, very little viral resistance to tenofovir has ever been reported, making it a popular option for first line treatment. In addition, side effects are uncommon and are usually mild when they do occur, including dizziness, nausea, rash and diarrhoea.
The main side effect limiting its use for longer-term treatment is kidney toxicity, similar (though milder) than adefovir to which it is chemically related. Patients on treatment require monitoring of kidney function and impairment usually improves once the drug is stopped. Tenofovir also has good activity against HIV making it the preferred option for patients infected with both HIV and HBV. It is now off patent and available from a number of generic suppliers.
TAF (trade name Vemlidy) is the most recent addition to treatments available for hepatitis B. It was developed to improve on the toxicity profile of TDF at a time when the patent of TDF was expiring. Like TDF, TAF is a pro-drug, converted in the body to its active form, tenofovir.
When compared directly with TDF, TAF has equivalent activity against hepatitis B with an improved toxicity profile (generally less kidney impairment on long-term treatment). However, the treatment has currently not been approved by NICE for treatment of HBV. It is likely that at this time, the improved safety profile of TAF compared to TDF would not justify the significantly higher costs and TAF is not currently recommended for first line treatment of HBV-infected individuals in UK. Like TDF, TAF has good activity against HIV and has been co-formulated into a number of HIV treatment options. For patients with HIV co-infection unable to take TDF, guidance has been developed to identify patients who can access TAF.
Treatments for hepatitis D are very limited and it is recognised as an important area where further drug development is needed, as patients infected with HBV and HDV tend to have more rapid progression of liver disease.
Pegylated interferon (covered elsewhere in more detail) remains the mainstay of treatment and prolonged courses (48 weeks) of interferon-based treatment, with the associated high rates of adverse events, can achieve sustained suppression of HDV in only a minority of patients. Drugs in development (such as bulevirtide) that may be used in combination with interferon, look promising in improving treatment outcomes, but further trials are still needed.
Hepatitis C (HCV) treatment has transformed since the discovery of the virus in 1989. Since 2014 progress has been particularly rapid in what has been one of the greatest changes in medicine of the 21st century to date. In contrast to treatment for HBV, treatment for hepatitis C aims to be curative. It is not possible to confidently predict who will be cured whilst taking treatment. Patients are monitored with blood tests and if no virus is detectable 12 weeks after finishing treatment they are said to have a “sustained virological response” (SVR12), which is the most commonly reported outcome. SVR12 is widely accepted as a surrogate outcome for long-term clinical benefits, although this has not been without some controversy (see section 15.18).
In broad terms, the evolution of treatment has moved from the interferon era (1991-2011), through a period when interferon treatment was combined with the first generations of direct-acting antivirals (DAAs, 2011-16), through to today when interferon-free DAA therapy is the standard of care. Through this period treatment has moved from requiring prolonged courses (24-48 weeks), using drugs with significant toxicity profiles (PEG-IFN and RBV, see below) giving poor outcomes, to shorter (8-12 week) therapy, with much improved side effect profiles and better outcomes (see Figure 15.13b).
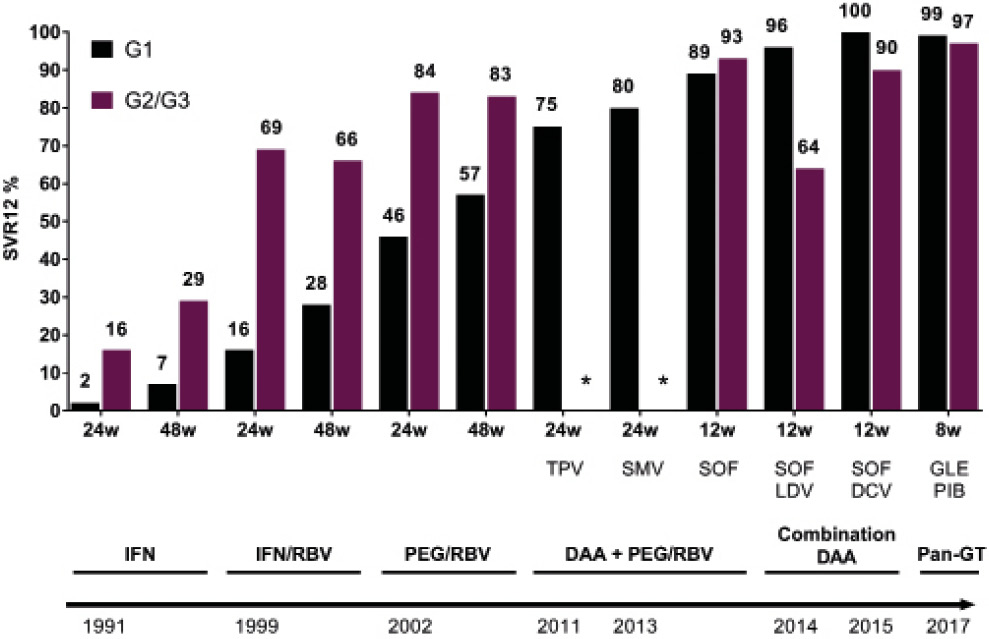
Figure 15.13b Estimated SVR12 rates from HCV treatment from 1991 to date. Shown by viral genotype; G1 or G2/3. IFN=interferon. PEG= pegylated interferon. RBV=ribavirin DAA=direct-acting antiviral. TPV=telaprevir SMV=simprevir SOF=sofosbuvir LDV=ledipasvir DCV=daclatasvir GLE=glecapreivr PIB=pibrentasvir (figure by Dr Chris Jones).
As with hepatitis B (above), the first treatments for hepatitis C were based on interferon, a naturally-occurring molecule which the body makes to fight infection and which can generate ‘flu-like’ symptoms when used as a medicine. This approach of strengthening the body’s natural response to treatment contrasts with current treatments directly targeting the virus (hence known as direct acting antivirals, DAAs). Throughout the 1990s and 2000s there were improvements in treatment, with the development of a different form of interferon (PEGylated interferon) and the addition of a drug which targets the virus (ribavirin).
PEGylated interferon is a modified form of the drug where a large polyethyleneglycol (PEG) molecule is attached. The advantage of “PEGylation “ is that drug levels in the patient stay higher for a longer period, reducing the need for dosing from 3 times/week to weekly, improving efficacy and reducing adverse events. PEG-interferon was approved by the FDA in 2002 and, where interferon is still used, remains the preferred treatment choice. Two versions were widely used – PEG-IFN-2b (trade name PEGintron) and peginterferon alfa-2a (trade name Pegasys). Despite improvements with PEGylation, the side effects of prolonged interferon use remained a major barrier to many patients completing treatment and for many, knowledge of the adverse events was a deterrent to starting therapy.
There is a long list of contraindications to PEG-interferon including: potential hypersensitivity to the active substance; autoimmune hepatitis; severe liver dysfunction or decompensated cirrhosis of the liver; severe pre-existing cardiac disease, including unstable or uncontrolled cardiac disease in the previous six months; HIV-HCV patients with cirrhosis and a Child-Pugh score ≥ 6 (see Q15.11) combination with telbivudine (see above); neonates and young children up to 3 years old; and in paediatric patients; the presence of, or history of severe psychiatric condition, particularly severe depression, suicidal ideation or suicidal attempt.
Given the widespread use of interferon and its associated toxicity, the adverse events are reproduced here in more detail than for other drugs (Table 15.13a) as a common challenge in practice is determining the extent to which symptoms experienced by individuals after cure can be attributed to treatment.
|
Body system |
Very common (≥1 in 10) |
Common (≥ 1/100 to < 1/10) |
Uncommon (≥1/1,000 to < 1/100) |
Rare |
Very rare |
Frequency not known |
|
Infections and infestations |
Bronchitis, upper respiratory infection, oral candidiasis, herpes simplex, fungal, viral and bacterial infections |
Pneumonia, skin infection |
Endocarditis, otitis externa |
|
Sepsis |
|
|
Neoplasms benign and malignant |
Hepatic neoplasm |
|||||
|
Blood and lymphatic system disorders |
Thrombo-cytopenia, anaemia, lymphadenopathy |
Pan-cytopenia |
Aplastic anaemia |
Pure red cell aplasia |
||
|
Immune system disorders |
Sarcoidosis, thyroiditis |
Anaphylaxis, SLE Rheumatoid arthritis |
Idiopathic or thrombotic TCP |
Liver and renal graft rejection, Vogt-Koyanagi-Harada disease |
||
|
Endocrine disorders |
Hypothyroidism, hyperthyroidism |
Diabetes |
Diabetic ketoacidosis |
|||
|
Metabolism and nutrition disorders |
Anorexia |
Dehydration |
||||
|
Psychiatric disorders |
Depression*, anxiety, insomnia* |
Aggression, mood alteration, emotional disorders, nervousness, libido decreased |
Suicidal ideation, hallucinations |
Suicide, psychotic disorder |
|
Mania, bipolar disorders, homicidal ideation |
|
Nervous system disorders |
Headache, dizziness*, concentration impaired |
Syncope, migraine, memory impairment, weakness, hypoaesthesia, hyperaesthesia, paraesthesia, tremor, taste disturbance, nightmares, somnolence |
Peripheral neuropathy |
Coma, convulsions, facial palsy |
|
Cerebral ischaemia |
|
Eye disorders |
|
Vision blurred, eye pain, eye inflammation, xerophthalmia |
Retinal haemorrhage |
Optic neuropathy, papilloedema, retinal vascular disorder, retinopathy, corneal ulcer |
Vision loss |
Serous retinal detachment |
|
Ear and labyrinth disorders |
|
Vertigo, earache |
Hearing loss |
|
|
|
|
Cardiac disorders |
|
Tachycardia, oedema peripheral, palpitations |
|
Myocardial infarction, congestive heart failure, cardiomyopathy, angina, arrhythmia, atrial fibrillation, pericarditis, SVT |
|
|
|
Vascular disorders |
|
Flushing |
Hypertension |
Cerebral haemorrhage, vasculitis |
|
Peripheral ischaemia |
|
Respiratory, thoracic and mediastinal disorders |
Dyspnoea, cough |
Dyspnoea exertional, epistaxis, nasopharyngitis, sinus congestion, nasal congestion, rhinitis, sore throat |
Wheezing |
Interstitial pneumonitis including fatal outcome, pulmonary embolism |
|
Pulmonary arterial hypertension |
|
Gastrointestinal disorders |
Diarrhoea*, nausea*, abdominal pain* |
Vomiting, dyspepsia, dysphagia, mouth ulceration, gingival bleeding, glossitis, stomatitis, flatulence, dry mouth |
Gastrointestinal bleeding |
Peptic ulcer, pancreatitis |
|
Ischaemic colitis, tongue pigmentation |
|
Hepato-biliary disorders |
|
|
Hepatic dysfunction |
Hepatic failure, cholangitis, fatty liver |
|
|
|
Skin and subcutaneous tissue disorders |
Alopecia, dermatitis, pruritis, dry skin |
Psoriasis, urticaria, eczema, rash, sweating increased, skin disorder, photosensitivity reaction, night sweats |
|
|
Stevens-Johnson syndrome, toxic epidermal necrolysis, angioedema, erythema multiforme |
|
|
Musculoskeletal and connective tissue disorders |
Myalgia, arthralgia |
Back pain, arthritis, muscle weakness, bone pain, neck pain, musculoskeletal pain, muscle cramps |
|
Myositis |
|
Rhabdo-myolysis |
|
Renal and urinary disorders |
|
|
|
Renal insufficiency |
|
|
|
Reproductive system and breast disorders |
|
Impotence |
|
|
|
|
|
General disorders and administration site conditions |
Pyrexia, rigors*, pain*, asthenia, fatigue, injection site reaction*, irritability* |
Chest pain, influenza like illness, malaise, lethargy, hot flushes, thirst |
|
|
|
|
|
Investigations |
|
Weight decreased |
|
|
|
|
|
Injury, poisoning and procedural complications |
|
|
|
Substance overdose |
|
|
Table 15.13a Adverse events associated with pegylated interferon (including when used with ribavirin) for HCV treatment. Those marked with * have been associated with treatment for HBV. Adapted from SPC for Pegasys www.medicines.org.uk/emc/product/1697/smpc
Ribavirin is a prodrug, a nucleoside analogue of guanosine. Although its effects on HCV are likely to be, in part, a consequence of inhibiting the HCV viral polymerase enzyme, its mechanisms of action remain the subject of some debate (for example it also appears to induce mutations in the virus which limit the virus’ ability to replicate).
The main challenge of using ribavirin therapy, particularly for long periods, is toxicity. Until recent years ribavirin was only used together with interferon for the treatment of hepatitis C and establishing which of the medications was responsible for individual side effects could be challenging. The most common challenge specific to ribavirin is anaemia, resulting from the breakdown of red blood cells (haemolysis). Other very common side effects include neutropaenia (a low level of the white cells-neutrophils), depression, insomnia, headache, dizziness and impaired concentration. There is a long list of common side effects, including thyroid disorders, mood alteration, emotional disorders, anxiety, aggression, memory impairment, visual blurring, vomiting, rash, photosensitivity back pain, impotence and weight loss. The drug is contraindicated in pregnancy due to risk of foetal abnormalities and male partners of women wishing to become pregnant should also avoid the use of ribavirin. It is also contraindicated in those who may have hypersensitivity, women who are breast-feeding, and individuals with severe cardiac disease or haemoglobinopathies (such as sickle cell disease).
In general, ribavirin still has a role in a limited number of patients for treatment, though as other treatments have improved it has begun to fall out of favour.
A major change came in 2011 with the approval of two new drugs, telaprevir and boceprevir, followed soon by simeprevir. These were the first direct-acting antivirals (DAAs) designed to target specific genes important for the survival of the viruses (and all targeted the protease, hence known as protease inhibitors). These drugs were prescribed with interferon (and ribavirin) and significantly improved SVR12 rates. However, there were additional toxicities from this approach and the drugs only targeted certain genotypes of virus. This has led them to fall out of favour and none are currently available in the UK. Sofosbuvir, first approved in the UK in 2015, has become a key treatment in current recommendations.
Boceprevir (trade name Victrelis) is an oral medication that was used in combination with interferon/ribavirin. It is an inhibitor of the HCV protease (“protease inhibitor, or PI”). It only has activity against genotype 1 infection, the most common genotype of infection in the UK. Very common side effects included anaemia, neutropaenia, anorexia, anxiety, depression, insomnia, irritability, dizziness, headache, diarrhoea, nausea, vomiting, rash, alopecia, dry skin, arthralgia and myalgia. It was contraindicated for use in those with likely hypersensitivity, pregnancy, autoimmune hepatitis and with a range of medications that interact with it. Its limited activity against all genotypes, toxicity profile and advances in other therapies led to it being withdrawn from the market in 2018.
Telaprevir (trade name Incivio) is an oral medication that was used in combination with interferon/ribavirin. It is an inhibitor of the HCV protease (“protease inhibitor, or PI”). It only has activity against genotype 1 infection, the most common genotype of infection in the UK. Very common side effects included anaemia, diarrhoea, nausea and vomiting with pruritis (particularly perianal itching) a common complaint. Rashes were very common with telaprevir, but could be very serious and specific guidance on managing rash was part of the product information. The medication was contraindicated for use in those with likely hypersensitivity, pregnancy, autoimmune hepatitis and with a range of medications that interact with it. Its limited activity against all genotypes, toxicity profile and advances in other therapies led to it being withdrawn from the market in 2018.
Simeprevir (trade name Olysio) is an oral medication that was used in combination with interferon/ribavirin. It is an inhibitor of the HCV protease (“protease inhibitor or PI”). It only has activity against genotype 1 and 4 infections. Due to its use with PEG-IFN/RBV it was not used in pregnancy. More commonly reported side effects (when not used with IFN/RBV) included constipation, sensitivity to sunlight and rash (which was usually mild, but could be severe). Simprevir was withdrawn from the UK market in 2018.
Sofosbuvir transformed the field of hepatitis C treatment. Developed by the company Pharmassett (as PSI-7977), it was acquired by Gilead Inc when they bought that company for $11.2 billion in 2012. Its initial price tag of $84,000 a course (£35,000 in the UK) made the drug a high profile example of pricing that was unaffordable for many health systems and the subject of a US senate enquiry. Prices have since fallen considerably, probably by over 90% in the UK, but the price paid by the NHS is generally considered commercially confidential. Sofosbuvir is central to a number of combination treatments now widely used for treatment of hepatitis C. It differs from the drugs above in that it targets the NS5B polymerase and has a low risk of resistant virus emerging. It has good activity against the range of HCV genotypes. When it was first used it was combined with PEG-IFN and RBV. Common adverse events included fatigue, headache, nausea and insomnia. Now it is most commonly part of an interferon-free combination (see below). No common adverse events are associated with sofosbuvir alone. On rare occasions a very low heart rate has been reported in patients with severe heart disease associated with sofosbuvir based combinations. One common phenomenon seen with all newer treatment given to patients with HBV and HCV is the reactivation of HBV once HCV is cured.
Four interferon-free sofosbuvir combinations have been prescribed in the UK, three of which remain part of the recommended care in 2020. The combination of sofosbuvir with other agents (ledipasvir, velpatasvir, daclatasvir and voxilaprevir) improved SVR12 rates and ensured high treatment rates were achievable without the additional use of interferon.
Sofosbuvir and ledipasvir (SOF/LDV, trade name Harvoni) is widely prescribed for treatment of HCV, particularly genotype 1 and there is relatively little data for genotype 3. Ledipasvir is an inhibitor of the NS5A. It is usually prescribed for 12 weeks and is approved for genotypes 1,4,5 and 6. In certain patients with mild disease high cure rates can be achieved with 8 weeks treatment. Severe side effects are uncommon, but headache, rash and fatigue are common in patients given sofosbuvir/ledipasvir compared to a placebo.
Sofosbuvir and daclatasvir (SOF/DCV not available as branded combination in UK) is a highly effective treatment combination for HCV genotypes 1-6 and is recommended by the WHO as a first line treatment option. It was the first treatment combination to show “pan-genotypic activity”. It is available at relatively low cost in low income countries. However, as different companies hold the intellectual property in developed markets its use is minimal outside of low income countries. Daclatasvir is contraindicated with a number of drugs that reduce its levels in the body and is not recommended during pregnancy or breast-feeding. SOF/DCV is generally well tolerated; common side effects include insomnia, headache, dizziness and migraine.
Sofosbuvir and velpatasvir (SOF/VEL trade name Epclusa) pairs sofosbuvir with a different drug of the same family as ledipasvir. Velpatasvir was developed to have a broader range of activity against genotypes 1-6 of infection, particularly genotype 3 (unlike ledipasvir) and one of the first “pan-genotypic” treatments. SOL/VEL is very well tolerated, only 0.2% of patients stopped treatment due to adverse events. The most common side effects (headache, fatigue and nausea) were no more common than in patients receiving a placebo.
Sofosbuvir, velpatasvir and voxilaprevir (trade name Vosevi) includes a third class of drugs (a protease inhibitor). It is active against genotypes 1-6 and approved for use with 8 weeks of treatment in patients without cirrhosis. It is generally recommended for patients who have failed a first course of DAA therapy and can successfully treat many patients who have developed resistant HCV. It is very well tolerated and only <0.1% patients discontinued treatment for adverse events. The most common adverse events include headache, diarrhoea, nausea, abdominal pain, vomiting and muscle pain. Its use is contraindicated with a range of drugs that can lower blood levels.
Elbasvir and grazoprevir (trade name Zepatier) is a combination of drugs targeting the protease and the NS5A of the virus. It is effective against genotypes 1,4,5,6 (it is not recommended for patients with genotype 2 or 3 infection). Treatment is 12-16 weeks and may be prescribed with ribavirin. Very common side effects reported include fatigue and headache; other common side effects include itching, nausea, abdominal pain, itching, joint and muscle pains and loss of appetite. Its use can be tailored using tests for resistant virus before treatment starts.
Glecaprevir and pibrentasvir (trade name Maviret) is a combination active against genotypes 1-6 of HCV infection and is licensed for 8 weeks of therapy, shorter than most other available combinations. It is contraindicated for use with a wide range of drugs which reduce its levels in the body and with patients with advanced cirrhosis. It is generally well tolerated, with only 0.1% discontinuing due to adverse events. Common adverse events include headache, diarrhoea, nausea, itching and fatigue.
The preferred treatment options for hepatitis C are handled differently in the home nations of the United Kingdom. In general, one of three treatment options is preferred; glecaprevir/pibrentasvir, elbasvir/grazoprevir or sofosbuvir/velpatasvir for initial treatment, with sofosbuvir/velpatasvir/voxilaprevir reserved for retreatment. Cost has been a major limiting factor in availability of treatment until recently, but in general, patients requiring treatment are now able to access it freely.
The response to this question will focus on 15.14 (a), 15.14(b) is addressed within the answer to 15.15
Pegylated interferon treatment cannot be used in patients who have cirrhosis of the liver. Treatment is most likely to be successful in patients with high levels of inflammation (assessed using a blood test for alanine transaminase (ALT)), moderate levels of virus and infection with HBV genotypes A or B (see also Q15.13).
Virtually all patients treated with tenofovir or entecavir respond to treatment. In patients who already have cirrhosis when they start treatment, the liver fibrosis (scarring) may regress in the majority, unless they are overweight and have fatty liver disease in addition to HBV infection. In patients with chronic HBV infection there is always a risk of developing liver cancer. In patients with cirrhosis this risk is substantially reduced, but in patients who do not have cirrhosis, the increased risk of cancer is virtually eliminated.
As outlined in 15.13, the treatment of hepatitis C has transformed dramatically since 2014. The predictive factors for treatment success have been similar over time, but are best considered separately within the interferon era, and the interferon-free era.
For interferon-based therapy, viral genotype was one of the most important factors in determining outcome. In general, genotypes 2 and 3 (including subtypes) could be treated with shorter courses (24 weeks) of therapy, whilst genotypes 1 and 4 required longer courses of therapy (up to 48 weeks).
In addition, the presence of cirrhosis was strongly predictive of response to treatment, with significantly lower SVR rates and more adverse events.
Other factors of note included gender (better responses for women), age (better responses if younger), viral load (better responses with lower viral load at baseline), previous treatment with interferon (results better if not previously treated) and human genetics. Individuals with the IL28B CC genotype responded better to IFN-based treatment, a rare example in modern medicine of where genetic testing was sometimes used to support therapeutic decisions.
In general, licensed courses of DAA therapy are able to achieve high SVR rates (>95%) in most patients, even if some of the negative predictive factors (above) were present. Cirrhosis remains important in determining duration of therapy required (and to a lesser extent, choice). A lack of previous treatment and low baseline viral load also predict a better response to therapy. The impact of viral genotype and viral resistance depends on the choice of treatment being used, with the newer pan-genotypic treatment (SOF/VEL, SOF/VEL/VOX, GLE/PIB) being able to achieve high cure rates across genotypes.
This question is addressed above in Q15.13
As noted in more detail in response to question Q15.22, establishing a causal relationship between a specific treatment and symptoms can be challenging in an individual patient, particularly when that condition is relatively common in the general population. However, there are many adverse events that have been associated with treatment, particularly interferon and ribavirin, and those in Annex 2 will be considered in respect of the Summary of Product Characteristics (see Table 15.13a).
Hepatitis B treatments have generally been well tolerated with minimal short or long-term adverse events. Impairment of renal function due to adefovir or tenofovir based treatments can persist after treatment finishes, although usually it is reversible. Loss of phosphate through the kidney associated with long-term use can lead to bone thinning (osteoporosis) with an increased risk of fracture.
Direct-acting antivirals for hepatitis C have not been associated with common or long-term adverse events, with the rare exception of skin rashes which can be severe.
The greatest challenges have been following prolonged treatment with interferon and ribavirin. These are addressed more fully in response to question Q15.13.
In respect of Annex 2, most conditions listed are reported in the summaries of product characteristics (SPC) for interferon (see above). Specifically mentioned are mood and cognitive disorders, digestive difficulties, fever and ‘flu-like’ symptoms, hot flushes, sepsis, vascular effects, cardiac arrhythmias, hypothyroidism, fatigue, headaches, dizziness, hearing loss, skin conditions, arthritis, alopecia, oral candidiasis (thrush), visual problems, impotence, coughs, pulmonary fibrosis, wheezing (asthma). Avascular necrosis is not listed, but there is some limited literature associated with HCV and/or interferon use.113
In general, interferon does not cross the barrier between blood and the brain well.
In monkeys, experiments with PEG-linked drugs have shown an association with “vacuolation” within the choroid plexus of the brain, suggesting direct damage to these cells within the brain.114 At high doses, significant changes were observed under a microscope, but there were no overt signs of effects on brain function.
The clinical relevance of these findings is not known. In general, there have not been significant differences noted in the toxicity from PEG-interferon compared to older, PEG-free interferons (both have significant toxicity from the interferon component).115 In addition, PEG doses used are relatively low, making toxicity less likely.116
The natural history of hepatitis B is altered by simultaneous infection with HIV. Immune control of HBV is negatively affected leading to a reduction of HBs-antigen seroconversion. If HBV persists, the HBV DNA levels are generally higher in HIV-infected patients not on antiretroviral therapy. With progression of cellular immunodeficiency (falling CD4 counts) reactivation may occur despite previous HBsAg seroconversion. However, after immune recovery resulting from anti-HIV therapy, HBeAg and HBsAg seroconversion do occur in a higher proportion of HBV/HIV patients, compared to seroconversion rates in HBV mono-infected patients treated for chronic hepatitis B.
In the untreated HIV-infected population, faster progression to liver cirrhosis is reported for HBV/HIV co-infected patients. Moreover, hepatocellular carcinoma may develop at an earlier age and is more aggressive in this population. Being HBV co-infected results in increased mortality for HIV-infected individuals, even after the introduction of highly active antiretroviral (combination) therapy (HAART). The EuroSIDA Study showed a 3.6-fold higher risk of liver-related deaths among HBsAg-positive patients compared to HBsAg-negative individuals.117 In the Multicentre AIDS Cohort Study (MACS), an 8-fold increased risk of liver-related mortality was seen among HBV/HIV-co-infected, compared those with HIV alone.118 Improved treatments for HBV have improved the outcomes for patients with HIV/HBV co-infection, but even with the widespread use of tenofovir (see Q15.13), HBV/HIV co-infection is still associated with an increased morbidity119 and liver-related deaths in HBV/HIV-infected patients still occur.
In the setting of HIV co-infection, the likelihood of spontaneous clearance of HCV infection decreases substantially and is below 10-15%. With progressive immunodeficiency (low CD4 cells) the risk of dying from HCV- associated liver disease increases significantly.120 HIV co-infection has been demonstrated to accelerate fibrosis progression. Although successful antiviral combination therapy for HIV and subsequent immune reconstitution can slow down the faster fibrosis progression, risk of hepatic decompensation (see Q15.11) still remains higher in HIV/HCV co-infected individuals on anti-HIV treatment compared to HCV mono-infected subjects.121 Sustained virological response (SVR) or cure after interferon (IFN) and ribavirin (RBV) combination therapy tended to be significantly lower in HIV co-infected individuals, particularly in genotype 1 patients. Therefore, previous EACS guidelines recommended longer treatment durations of up to 72 weeks for co-infected genotype 1 and 4 patients who still had a detectable HCV-RNA 4 weeks after starting HCV therapy and at least a >2log drop in HCV-RNA at week 12 of HCV therapy.122 Treatment for GT2 and 3 could last up to 48 weeks. Considering the longer HCV treatment durations, a higher risk for cumulative IFN and RBV-related toxicity existed for HIV co-infected individuals. In the era of all oral DAA combination therapy similar SVR rates have been observed for HIV co-infected and HCV mono-infected subjects.123
Both hepatitis B and hepatitis C are more common in people living with HIV (compared to the general population) and advanced HIV is associated with a worse outcome for both viruses. However, they are not considered opportunistic infections, nor are they part of the syndromic diagnosis of AIDS.
The virus responsible for hepatitis A infection is highly contagious and is transmitted primarily by water or food contaminated with faecal matter. Rarely, blood transfusions and clotting factor concentrates have transmitted hepatitis A. In 1994 a report was published where hepatitis A was transmitted by a factor VIII concentrate treated by a virucidal method (solvent-detergent) that ineffectively inactivated non-enveloped viruses.124 Hepatitis A is a self-limited infection that only lasts a few weeks for most patients and there is no ongoing carrier state.
As hepatitis C virus (HCV), hepatitis B virus (HBV) and hepatitis D virus (HDV) are transmitted via similar routes (including through blood or blood products), dual infection and even triple infection can occur in some patients at the same time. In dual infection with HBV and HCV, as well as in triple infection with HBV, HCV and HDV, there is interference between the viruses where one virus might suppress the others. Of particular clinical relevance is the re-emergence or flare of HBV following successful treatment of HCV.
Whether HBV/HCV co-infection increases the risk of liver cancer is complicated by the balance between the direct effects of each virus and the interaction between them (for example, individuals with HBV who become infected with HCV will have a higher risk of cancer due to HCV, but also a lower HBV viral load which reduces the risk from HBV).125
Outcome of triple infection with HBV, HCV and HDV has been reported to be more severe in the acute super-infection stage.126 Moreover, infection with HBV, HCV and HDV increases the risk of liver failure. Patients with triple infection of HBV, HCV and HDV also have a higher risk for cirrhosis development and an increased risk for progression to HCC.
Hepatitis D is the least common type of chronic viral hepatitis. The hepatitis D virus is a defective RNA virus (see Q15.2) that replicates only in the presence of hepatitis B surface antigen (HBsAg). Individuals infected with both HBV/HDV can progress more rapidly to cirrhosis when compared to those with HBV alone. Treatment for HDV is limited (see 15.13).
Hepatitis G is caused by a small RNA virus with similarities to HCV. The GB virus C/GBV-C has now been identified as a human pegivirus. The virus, although it causes persistent viraemia for at least six months and often many years, does not cause appreciable clinical symptoms. However, jaundice, fatigue, nausea and loss of appetite can occur in the acute phase. Although HGV is a co-infection for many individuals with chronic HCV, it does not alter the clinical course of HCV, nor interfere with the response of HCV to interferon treatment. It is clearly associated with transmission by transfusions. Mild elevations of ALT and, rarely, jaundice have been seen in transfusion recipients, but none were found to have chronic liver disease associated with HGV alone. About one third cleared the virus within three years of becoming infected with HGV and infection with HGV did not alter the course of chronic hepatitis C. 127,128
There have been several reports associating GBV-C with a better outcome of HIV infection. In general, GBV-C/HIV co-infection is associated with lower HIV viral load, slower progression to AIDS and improved response to HIV treatment. These effects are more likely to be seen with advanced HIV infection (low CD4 counts).129 Theoretically, in HIV/HCV/GBV-C triple-infected individuals, treatment of HCV with interferon could also lead to clearance of GBV-C. This may, paradoxically, worsen the outcome of HIV infection. However, this was not supported by clinical studies.
Human pegivirus 2 (HPgV-2) is a novel blood-borne infectious virus of humans and is likely transmitted via the parenteral route.130 HPgV infection is considered non-pathogenic.
Human parvoviruses can be transmitted parenterally and they have been identified within blood products.131,132 Persistent infection with PARV4 and B19 viruses has been reported. Reports of an association between HBV and PARV4 infection have been inconsistent and there is no evidence that co-infection affects prognosis. One study of PARV4/HIV/HCV infection found no evidence of an effect on HCV outcomes.133 A higher prevalence of parvovirus B19 and both HBV and HCV has been reported, but there is no clear evidence of an effect on HBV progression or severity.134
CMV is a common viral infection in the general population and there is little evidence of association with HBV or HCV infection, nor with outcomes of those infections. There is mixed evidence that CMV may contribute to severe HCV after liver transplantation.135
Multiple hepatitis C genotypes can infect one individual. Older tests of genotype may only detect the most common genotype present, but new methods of sequencing (“next generation deep sequencing, NGS”) are giving greater insight into the prevalence of multiple infections. The main clinical relevance relates to the choice of treatment, as different drug combinations may have different activity against genotypes of infection. It is well recognised that a genotype(s) present at low levels may re-emerge after successful treatment of other co-infecting strains,136 although with the use of pan-genotypic treatments this is less likely to be a problem. There is no evidence to suggest that infection with more than one genotype otherwise affects progression of disease or prognosis.
Please note that as, far as possible, section 15.16 contains responses to Supplemental
Question 14.
In the largest study of UK haemophiliacs, individuals treated with blood products between 1969 and 1985 were followed up until 1993. Deaths from liver disease were 16.7 times higher and deaths from liver cancer 5.6 times higher than in the general population.137 Most of this increased risk was probably due to hepatitis C infection. An excess of HCV-related deaths was also seen in a US study of transfusion-associated hepatitis; that is, in patients receiving transfusion for other indications.138
Von Willebrand’s (vWD) disease is less common than haemophilia and those affected tend to have significantly less exposure to pooled donor blood products.139 However, those with vWD who are infected with HCV appear to have similar incidence of liver disease and cancer to individuals with haemophilia.140
In patients with hepatitis C, iron overload may increase the risk of liver fibrosis. In thalassaemic patients with iron overload, it is possible that risks of HCV progression and cancer will be higher, but there is not good evidence to support this. There is no evidence of an increased risk of HCC in patients with HBV/thalassaemia.141
Because of the faster liver disease progression in HIV co-infected patients with HCV, many studies in patients with haemophilia, or other bleeding disorders, are confounded by the underlying HIV co-infection. Studies in HCV mono-infected haemophiliacs, however, were able to show a slow progression of hepatitis C and confirm that the natural history of HCV infection in these patients is not different from those without congenital bleeding disorders.142 In general, the genotype distribution in the HCV population has a wide ethnic and geographical variation. Therefore, in patients with bleeding disorders, the genotype distribution usually reflects the genotypes of the respective blood donor population. One important characteristic in this group of patients is that a significant proportion has a mixed-genotype infection due to multiple exposures to contaminated blood products over a period of years.143 Overall, HCV genotype 1 seems to be the dominant one.144
The antiviral treatment of HCV in patients with hereditary bleeding disorders is not different from that of any other infected patients. Nevertheless, many patients with hereditary bleeding disorders have declined (PEG)-interferon-based treatment because of side effects. Lower SVR rates have also been reported for IFN-based HCV therapy in haemophiliacs.145 In the DAA era, however, similar high HCV cure rates have been observed in patients with bleeding disorders.146
No data has been reported which suggests any difference in the natural course of hepatitis B in persons with hereditary bleeding disorders or in response to standard hepatitis B therapy with nucleos(t)ide analogue therapy. Patients with hereditary bleeding disorders who acquired hepatitis B and developed chronic hepatitis B are at increased risk for development of cirrhosis and development of hepatocellular carcinoma. Occult hepatitis B infection (OBI) is a form of chronic HBV infection characterized by low level HBV DNA without detectable HBV surface antigen (HBsAg).
OBI is frequently associated with the presence of anti-HBc and in some cases also with anti-HBs. Patients, who formerly received non-inactivated factor concentrates, can potentially be considered at high risk for OBI, especially as these patients usually are HIV or HCV co-infected. Studies assessing the rate of OBI found the incidence to be moderately low (1.8%).147 However, close monitoring of these infections is of clinical significance, especially in patients with co-infections and concomitant immunosuppression.
Although there is not strong evidence to suggest that infection with HBV or HCV per se affects bleeding disorders, it is important to note that (even in the absence of a specific bleeding disorder) patients with advanced liver disease/cirrhosis have problems with clotting (see also Q15.11). It is possible that in such individuals this may affect bleeding disorders due to low production of proteins in the liver.
Where patients with haemophilia (A or B) or von Willebrand’s disease have undergone liver transplantation, there is the potential for the transplant liver to improve production of clotting factors, though this may be influenced by the presence of inhibitors.148
The nature of chronic infection and treatment is fundamentally different between hepatitis B and hepatitis C. The aim of treatment for hepatitis B is to suppress viral replication, but the virus itself is usually not eliminated. For hepatitis C the aim of treatment is to cure infection.
The current treatments for hepatitis B suppress the replication of HBV and, by doing so, prevent ongoing damage to the liver with resolution of inflammation and, in some cases, improvement in scarring (fibrosis) due to the virus. However, for all patients there is still evidence of persistent infection (typically measured by detecting HBsAg “surface antigen” in the serum). When treatment is stopped the virus will usually return, though some patients do gradually clear infection over time and can come off treatment without a rebound in virus levels.
Tests to detect the presence of HBV DNA and/or HBsAg are used to assess whether the infection has been cleared from the blood. However, it is well described that individuals previously infected with hepatitis B can reactivate the virus if they are exposed to illness or treatment that suppresses their immune system. Guidelines now exist for the prevention of such reactivation (in patients undergoing chemotherapy, for example) by prescribing antiviral treatments.
The area of hepatitis B “cure” research is currently very active and will likely yield new treatments over the coming years (see more detail below in section 15.25).
As discussed in response to Q15.13, there are limited treatments available for hepatitis D, though this remains an area of active research and development. Prolonged courses of PEG-IFN are able to achieve a sustained virological response in a proportion of patients who can achieve an effective cure. Cure is defined as persistent absence of HDV RNA using a NAT test after completion of treatment. Relapse of HDV infection following complete viral suppression on treatment is common.
Hepatitis C does not integrate into the host DNA of an infected individual and the aim of treatment is cure. It is not possible to tell whether cure has been achieved until treatment has finished. If no virus is detected in blood after the end of treatment, a patient is said to have achieved a sustained virological response (SVR). In patients whose virus returns after unsuccessful treatment, it is usually detectable 12 weeks after treatment finishes and this is generally taken to be the time point where “cure” is defined. Historically, SVR24 was more commonly used, but reappearance of the virus between weeks 12 and 24 is rare. SVR is tested by detection of virus from a blood sample, usually using HCV RNA PCR or and HCV antigen test.
It is worth noting that reinfection with HCV after successful treatment is possible and previous infection does not confer significant protection against further infection. This means that when HCV becomes detectable after a long period of undetectability post-treatment, there needs to be an effort to determine whether the virus is the same as the original virus. New methods to sequence the genetic viral material are increasingly helpful to resolve this question.
When cure is defined in these terms it is important to recognize some areas of contention and/or confusion.
(i) SVR12/24 is a surrogate end-point for long-term clinical benefit
The objective of treatment is primarily to reduce long-term complications of infection and, in some cases, prevent the transmission of the virus to uninfected individuals. As discussed in Q15.11, progression of the disease (and regression) is slow and thus measuring virological cure at 12 or 24 weeks is a surrogate for longer-term improvement in health.
There is good evidence from placebo-controlled trials that on average, patients experience improved quality of life when on active treatment and that these changes persist after successful treatment.149 However, a patient may feel no immediate subjective benefit when SVR is achieved.
(ii) Absence of replicating HCV (undetectable virus in blood) does not mean a patient is cured of long-term consequences of previous HCV infection
There is a large body of evidence that achieving SVR12/24 is associated in the longer term with significant reductions in all cause mortality, liver cancer and liver failure.150 However, these risks remain higher than in patients never infected with HCV. Within the field there is broad consensus that SVR 12/24 can be considered a surrogate for long-term cure; however, this view is not universal. There have not been trials of interferon-free HCV treatment against placebo able to compare the difference in long-term clinical outcomes between those who achieve SVR and those who do not. Such trials have been proposed,151 though most experts would consider such a study unethical given what is already known about hepatitis C treatment. This debate came to the fore with widespread coverage of a review by the Cochrane group, which was reported as showing “no evidence of clinical benefit” of newer HCV treatment.152 However, the review only addressed studies with relatively short clinical follow-up. The strong push back from the HCV community at the time was, in part, because such arguments had the potential to limit access to treatment.
(iii) Cure of HCV, as defined by a lack of replicating virus in the serum, does not mean that no virus is detectable in the body
It is possible in some patients to detect the presence of RNA from hepatitis C virus despite achieving SVR, with the virus undetectable within plasma. This is likely to reflect fragments of viral material detectable within cells, but it does not imply that this is necessarily a viable virus. Cases of reappearance of HCV are very rare, sufficient to merit being written as case reports. In such cases it is important to rule out the possibility of reinfection and, even in the era of molecular genetics, it is not always possible to establish this beyond reasonable doubt.
After HBV infection a patient is likely to maintain antibodies to core (HBcAb) and surface “s” antigens (anti-HBsAb) and “e” antigen (anti-HBeAb). A test of clearance requires direct detection of virus, including a sensitive HBV DNA test, as well as detection of HBsAg. There are no tests validated to detect the clearance of cccDNA (see Q15.25).
After HCV infection patients maintain anti-HCV antibodies. Tests of cure require a sensitive HCV RNA detection method (usually PCR) or HCV antigen testing (less sensitive, but usually sufficiently sensitive to detect any ongoing infection).
Where acute infection is spontaneously cleared (as discussed in Q15.9), and the patient returns to normal health, there are no recognised long-term implications for health. Indeed, cleared infection may give some protection against further infection.
The long-term health consequences after chronic infection are most relevant for hepatitis C, as it is rare for chronic HBV to be cured with treatment. What follows assumes cleared means that the person previously chronically infected is now HCV PCR negative. Long-term consequences can result from (i) the consequences of viral damage to the liver, (ii) the consequences of viral damage outside the liver (“extrahepatic manifestations”), (iii) the long-term effects attributable directly to treatment, and (iv) the psychological impact of infection (not addressed here).
The major factor determining any long-term impact on a person’s health would be the degree of liver fibrosis at the time the HCV PCR became negative. This can be considered as those with cirrhosis (advanced fibrosis) and those with mild to moderate fibrosis without cirrhosis.
After cure, a person with cirrhosis would expect some regeneration of the liver, which would improve health and symptoms of liver failure, but they may be left with residual symptoms and signs of liver failure. This would be a small proportion of those with cirrhosis and such persons may require liver transplantation.
The majority of those with cirrhosis, who have never had liver failure, are likely to get some improvement in liver function following liver regeneration after SVR. Despite this, they still have a long-term risk of developing hepatocellular carcinoma. When infected with HCV and cirrhotic the rate of HCC development is 2-8% per annum; following SVR over 3 years this risk falls to less than 2%; evidence beyond this time frame is currently lacking. It is believed that the risk would continue to fall, but remain elevated compared to the general population.
The person with cirrhosis would also have the risk of having oesophageal varices and, as a consequence, the risk of a variceal haemorrhage (see 15.11). This risk should be reduced to very low in non-progressive liver fibrosis, that is, after the causal disease has been cured. Persons with varices would need to take long-term medication called beta blockers, or undergo regular endoscopy and oesophageal banding. Those without varices would require 3 yearly upper GI endoscopies to check for the development of varices.
Persons with cirrhosis or significant fibrosis would be at increased risk of the consequences of co-existing liver disease, most commonly non-alcoholic fatty liver disease or alcohol-related liver disease. If persons already have fibrosis due to HCV infection and then develop another liver disease, their prognosis will be worse because they need less additional fibrosis to cause significant liver health impacts.
The other impacts on long-term health beyond liver disease are covered below in the responses to Q15.20-22.
Hepatitis is an inflammation of liver cells, it is not specific to HBV or HCV. Many patients with hepatitis have no symptoms. If there is no longer hepatitis, then any remaining symptoms are not due to hepatitis. The hepatitis causes damage to the liver; usually all this damage is repaired. However, as described above in answer to 15.19, if there is significant fibrosis then the symptoms of liver impairment will be apparent. The answers to 15.21 and 15.22 deal with remaining symptoms that people may experience after HCV infection, including extra-hepatic manifestations of HCV infection.
To answer the question about post treatment complications or effects one must also consider any treatment side effects that persist. The answer is very dependent on what treatment has been used; the major distinction being interferon containing therapy and interferon-free therapy. It is important to note that for much of this area there is an absence of high quality evidence, rather than clear evidence of an absence of effect.
Interferon containing regimens have no predictable long-term effects that have been detected in the available published clinical trial follow-up data. This data shows no ongoing symptomatology different from controls, and improved quality of life with levels in those that achieved SVR returning to the normal range. This data has the power to detect events that are more frequent than 1 in 1,000, and that cause symptoms that were specifically asked about or are diagnosed during follow-up by other medical practitioners. On treatment, depressive side effects are well described with interferon; the studies that have looked at this have demonstrated resolution of depressive symptoms back to pre-treatment levels relatively rapidly after treatment, with no clear evidence of an effect within 12 months of follow-up. Interferon is an immune modulator and is associated with flares, or emergence, of predisposed autoimmune disease; this is at the level of case reports, so it is not possible to ascribe a rate, but would be less than 1 in 1,000 cases. Ribavirin has, for the most part, been used in conjunction with interferon and the two should be regarded as potentially co-responsible for any ascribed effect. Only small numbers of patients received ribavirin, without interferon, in combination with new DAA therapies, so the same limitations of data apply. Below is a list of conditions associated with interferon alpha therapy.
Hypo/hyperthyroidism, depression, fatigue, sarcoidosis, lichen planus, skin vasculitis, peripheral neuropathy, Bell’s palsy (see Table in 15.13a).
Treatment with non-interferon- based treatments has shorter follow-up periods and the reported data shows no evidence of any long-term problem, within the caveats explained under interferon regimens.
The psychological post treatment effects are very difficult to disentangle. The limited evidence shows improved quality of life across populations. For an individual patient experiencing psychological distress, it is often difficult to ascribe an association with treatment, with multiple other life events and concomitant factors being relevant in individual cases.
15.21 and 15.22 have been dealt with together due to overlap in the answer. The issues of treatment-related side effects and complications related to treatment have been dealt with in the answer to 15.20.
Dealing first with HBV, there is much less evidence of extra-hepatic manifestations as a consequence of HBV infection compared to HCV. In a one cross sectional study, 16% of patients153 had extra hepatitis symptoms, some combination of myalgia, arthralgia, arthritis or dry eyes. Much more rarely, patients can develop glomerulonephritis alone or as part of polyarteritis nodosa; these syndromes normally resolve with progression of the HBV or with resolution of acute infection.154
With regard to HCV, response to these questions has first been laid out in a list form for the most commonly recognised extra-hepatic manifestations of HCV infection, with a grading as to the strength of evidence. The conditions that follow have been grouped according to the body system or organ affected; where in Q15.22 Annex 3 specific conditions have been listed, these are responded to under the system or organ affected.
Conditions with significant prevalence, consistent pathogenetic data
Mixed cryoglobulinemia/cryoglobulinaemic vasculitis
B-cell Non-Hodgkin lymphoma (NHL)
Conditions with higher prevalence in HCV-infected populations compared to controls
Type 2 diabetes mellitus
Insulin resistance and metabolic syndrome
Glomerulonephritis
Renal insufficiency
Cognitive impairment
Depression
Cardiovascular disorders (for example, strokes or ischemic heart disease)
Sicca syndrome
Arthralgia/myalgia
Auto-immune conditions, including: rheumatoid arthritis, SLE, thyroiditis
Monoclonal gammopathies
Porphyria cutanea tarda
Lichen planus
Parkinson’s Disease
Gall stones
Irritable bowel syndrome
Conditions with possible association with HCV
Polyarthritis
Chronic polyradiculoneuropathy
Lung alveolitis
Conditions with anecdotal reports of association
Polymyositis
Dermatomyositis
Polyarteritis nodosa
Psoriasis
Mooren corneal ulcer
Erythema nodosum
Pancreatitis
This is unequivocally a complication of HCV infection, due to progressive fibrosis caused by the HCV infection. The complications are described under the responses to 15.11 and 15.19. From Annex 3, the following can be directly due to advanced liver disease: thrombocytopenia causing bruising and/or extensive nose and other bleeds, variceal bleeds, encephalopathy, enlarged spleen and changes to bone (osteopaenia and osteoporosis). It also includes an increased risk of liver cancer. Liver damage increases the risk of pulmonary emboli. Liver disease is associated with an increased susceptibility to viruses and infections, including shingles. Patients with liver disease and ascites are more likely to develop an umbilical hernia, but there are other causes for this. Liver disease reduces fertility generally (see supplemental questions), but there is no evidence of a HCV-specific effect. Glandular fever is not likely to be associated with HCV infection, but is more likely to be diagnosed because of investigation of liver damage.
There is evidence that inflammatory response to HCV infection could cause atheroma. Additionally, there is good evidence that HCV infection is associated with increased incidence of the metabolic syndrome, which manifests as cardiovascular disease, non-alcoholic fatty liver disease and type 2 diabetes. The risks of the metabolic syndrome persist after HCV cure. The complications of metabolic syndrome include hypertension, cerebral haemorrhage, vascular dementia, transient ischaemic attacks (TIA), heart disease, sleep apnoea and polycystic ovarian syndrome (PCOS).
HCV infection is a risk factor for developing liver cancer. The metabolic syndrome also carries an increased risk of cancer, including ovarian cancer, lung cancer, breast cancer and bowel cancer. The evidence for skin cancer association is not clear and the 3 different types have different risk profiles. A specific type of Non-Hodgkin lymphoma (marginal zone B-cell lymphoma) is directly associated with HCV and regresses with HCV treatment, suggesting that HCV cure would dramatically reduce the risk of developing this tumour. Head and neck cancer, usually associated with HPV virus, has also been associated with HCV.156
HCV infection is associated with arthritis, including rheumatoid arthritis and polyarthritis. In a large, prospective study of 1,614 HCV-infected patients, arthralgias were the most common complaint, with a reported prevalence of 23%. A symmetric, inflammatory polyarthritis, primarily involving small joints and resembling rheumatoid arthritis (RA), has been described in association with HCV infection.157 Rheumatoid factor may be present in up to 50–85% of these patients (higher than the general population); however, unlike RA, no erosive joint changes are noted and overall the prognosis is better. The presence of true RA coexisting with HCV infection is far less common. In the United States, the estimated prevalence of coexisting HCV infection and RA is 0.02%. The arthralgia associated with HCV infection may lead to a mistaken diagnosis of fibromyalgia.158 Carpal tunnel syndrome and peripheral neuropathies are associated with increased likelihood of HCV infection. Spinal stenosis is a multi-factorial condition and there is no evidence associating it with HCV. Sacral agenesis is a rare birth defect and there is no evidence associating it with HCV infection.
HCV infection causes immune cell dysfunction. This is most obviously manifest as cryoglobulinemia, which can cause multi-system damage. This most commonly affects the kidney (evidenced by glomerulonephritis) and sometimes leads to kidney failure. HCV infection is associated with Systemic Lupus Erythematosus (SLE), of which Hughes Syndrome, Antiphospholipid Syndrome (APS) and Raynaud’s syndrome are variants, components or symptoms. Autoimmune thyroid disease leading to thyrotoxicosis or hypothyroidism are also associated with HCV. Platelet problems leading to purpura (bruising) is caused by HCV, both by an effect of a scarred liver and by causing idiopathic thrombocytopenic purpura (ITP).
The HCV brain fog syndrome is a consequence of infection; it usually improves or resolves with treatment. Some have residual symptoms; this is often complicated by the co-existence of advanced liver disease, which can cause occult hepatic encephalopathy, which has similar symptoms or may be attributable to depression and/or anxiety related to having been infected. HCV infection is associated strongly with mental health conditions, including bipolar affective disorder, depression and anxiety, as well as drug and alcohol dependency. There is no evidence that this is causally linked to active HCV infection and the association is confounded, as these are risk factors for acquiring HCV infection (this route of HCV transmission is not relevant to the focus of the Infected Blood Inquiry).
HCV infection is associated with interstitial lung disease (ILD), which may be misdiagnosed as chronic obstructive pulmonary disease (COPD) or asthma. There is no evidence of association with bronchiectasis.
There is no evidence of association between HCV infection and the following: rosacea, epilepsy or bladder problems, including bladder erosion and incontinence. Eye problems related to inflammation and ulceration of front parts of the eye are associated with HCV via an immune mechanism, but early cataracts and glaucoma are not. Periodontitis may be associated with chronic liver disease, but does not appear to be associated with HCV. It is not clear medically what is meant by “stress related skin conditions”, but there are a number of skin conditions associated with increased anxiety, including acne excoriée, lichen simplex, papular and nodular prurigo, dermatitis artefacta and trichotillosis.
The rate of miscarriage and foetal abnormality in normal pregnancy is higher than the general public believe. Available evidence does not suggest that rates of foetal abnormality or miscarriage in pregnant patients infected with HCV are different from the background rate. However, the cohorts studied are small and many are confounded by concomitant drug use by the mothers. In the Irish Rhesus cohort there was no evidence of excess risk. There is no evidence of HCV producing specific syndromes of speech and language difficulties in children.
Explanation being given to patients and families regarding a diagnosis of hepatitis should be done with consideration of the complexity of the disease and treatments, breadth of transmission mechanisms, and the health literacy of the patient and family. Moreover, this is often in the context of a disease about which they may not have detailed knowledge and there is evidence of ongoing associated stigma and discrimination.
Testing for hepatitis is increasingly commonplace and more often being done routinely as part of any blood testing to assess abnormal liver function159 or in sexual health screening done in primary care.160 Given this, a reasonable knowledge base for any primary care clinician is expected, in order to facilitate initial discussion with a patient who is found to be positive for hepatitis B or C.
There must be a suitable environment, with adequate time given for such an explanation. It would be unusual to expect a primary care clinician to give more than an initial diagnosis and brief explanation. This is mindful of both the potential shock of the diagnosis, but also noting that only a very small portion of the information discussed will be retained beyond the diagnosis.161 A primary care clinician should also be mindful that most patients and families may consider looking online for further information, and so signposting to authoritative NHS or third sector resources should be considered.
In a secondary care situation, where further details would be shared, the current level of understanding should be established in a suitable environment, with time given for a fuller discussion. Different learning and health literacy needs should be catered for, including the use of pictorial explanation4 and supporting patient self-recording;162 this can increase understanding and retention of information.
Clinicians should make no assumptions on patient preference as to treatment options. Both Realistic Medicine163 and NHS England164 highlight the important of effective shared decision making to achieve optimal patient care. It is important to listen to the patient preferences, sharing all decision making, having ensured the patient has the information they need to make an informed choice in an equal partnership.
Signposting and providing further written information for a patient to consider after a more in-depth secondary care review is also important, given information retention issues which are exacerbated if the patient is anxious during a consultation.
An overview of current transmission, prognosis and treatment options is summarised in this table.
|
Hepatitis B |
Hepatitis C |
|
|
Transmission |
The virus is passed on via body fluids, including: blood, semen and vaginal secretions. The virus can survive for at least one week outside the body in dried blood. |
Hepatitis C is the most common form of viral hepatitis. It is highly infectious. The virus is passed on via blood. It can be spread by sharing drug injecting equipment, contaminated items, by some forms of sexual activity or via mother-to-child transmission. |
|
Prognosis |
The majority of people make a full recovery. Up to 5% become ‘carriers’ with chronic (long-term) infection, with a risk of developing cirrhosis, liver failure and carcinoma of the liver. Approximately 1% develop a serious potentially fatal illness. |
>90% of people can be cured by antiviral treatment. If untreated there is a risk of developing cirrhosis, liver failure and carcinoma of the liver. |
|
Treatment Options |
In most cases no active treatment is required. If any of the complications described above develop, the range of treatments available include antiviral medication, and if necessary, liver transplantation. |
Antiviral treatment should be offered. In the case of treatment failure or any of the complications described above, treatment can be tailored to the condition and may include liver transplantation. |
Table 15.23a
If appropriate, the following information should be provided to newly infected individuals
An example of a patient information leaflet can be found here: (https://www.kch.nhs.uk/Doc/pl%20-%20820.1%20-%20chronic%20hepatitis%20b%20virus%20(hbv).pdf)
If appropriate, the following information should be provided to newly infected individuals
An example of a patient information sheet can be found here: (https://www.uhb.nhs.uk/Downloads/pdf/PiHepatitisC.pdf)
Neither HBV nor HCV infection should now be a barrier to starting a family, including through assisted conception. An infected adult (male or female) planning a family should seek medical assessment and advice, and may benefit from starting treatment, both for their own health and to prevent transmission to their partner and/or children. All patients benefit from pre-conceptual advice with regard to lifestyle choices and general health. This is particularly so if there is a pre-existing illness.
The general advice for anyone infected applies to the above, but key is ensuring sexual partners are protected from HBV by effective vaccination.
Pregnant women in the UK should be tested for HBV in pregnancy as part of routine antenatal care. A pregnant mother with HBV may be offered treatment during pregnancy to reduce the risk of transmission, and the newborn baby should be offered immediate vaccination, potentially also with specific immunoglobulin (antibody) treatment to prevent infection.
In general, the risk of transmission through breast milk is very low and mothers can breastfeed their children once the infant has been vaccinated.
An example of a patient information sheet tailored to mothers diagnosed with HBV in pregnancy can be found here:
Hepatitis C infection may be passed to the baby, although the risk is lower than with hepatitis B or HIV (see Q15.5). Although there is no effective vaccination for HCV, transmission via vaginal sex is rare. Where possible, treatment of an infected individual (mother or father) should be completed before conception.
HCV is not routinely tested as part of antenatal care in the UK, but testing is recommended for those considered at high risk.
This will require discussion and counselling in those contemplating pregnancy.
Although it is likely that HCV treatment in pregnancy is safe and effective, there is not enough evidence to support its use at the current time.
Caesarean section is not routinely recommended solely due to HCV infection. The risk of transmission via breastmilk is very low and breast-feeding should be encouraged (unless nipples are cracked or bleeding).
An example of a patient information sheet regarding HCV and pregnancy can be found here:
(https://www.hep.org.au/wp-content/uploads/2017/09/Factsheet-Hep-C-pregnancy-children.pdf)
Specific recommendations with regard to approaches for assisted conception are beyond the scope of this report.
Patients who have ever tested positive for HBV or HCV should not donate blood, irrespective of whether they have been symptomatic or not.165 All donated blood is routinely tested for hepatitis B, C and E, as well as HIV.166
The clinician-patient relationship is very important at any time, but is particularly so for patients suffering from long-term illnesses that are likely to have significant impact on the patient and their family. They require timely access to professional care from trusted teams to maintain support and continuity. They should be assured of not just care, but also understanding, respect and excellence.
The GP and primary care team are likely to be involved with the initial presentation and diagnosis of illness and any onward referral, but will also often be called upon for support with the psychological and social aspects of care for the patient and their families.
Throughout the course of the illness, this will often involve the provision of information that may simply be oral, but may also include appropriate written material or increasingly, signposting to relevant trusted websites or specific patient support groups and networks. It will include the management of test results, medication and referrals, both within the community and to secondary care.
Patients should know how the information about their hepatitis diagnosis is shared. The sharing of such information between health professionals is often critical for safe prescribing, given the common interactions with drugs used to treat hepatitis.
Referral to specialist care will involve many people and, while each of them may have specific roles in the management of the patient’s care and treatment, they are also likely to have shared roles and responsibilities. Good communication between everyone involved is essential if the patient is to have confidence in their care.
Increasingly, with the drive towards personalised medicine and shared decision making, it is important for patients to develop a knowledge of the natural course of their condition. In this way they will know what to look out for and when to seek advice or help, and be better able to manage the many effects of their illness on their daily lives.
The follow-up for HCV-infected patients after SVR will be tailored to an individual patient’s clinical condition and personal circumstances. Key factors in this will include whether the patient has evidence of liver fibrosis/cirrhosis, whether the patient is considered at risk of HCC, whether the patient is at risk of reinfection, and the personal preferences of the patient and their ability to attend clinics.
In general, a patient without evidence of liver scarring and no other co-morbidities likely to affect the liver (examples include high alcohol intake and obesity) may be discharged from specialist care.167 Those with significant fibrosis or cirrhosis are likely to require lifelong surveillance for the risk of HCC, with ultrasound scans of the liver and +/- AFP (alpha-feto protein) blood tests every six months (see also Q15.11).
In those considered at high risk of reinfection, a PCR or antigen test should be considered every 6 months to ensure early detection of a new infection (antibody tests will remain persistently positive from previous infection).50 Patients known to have varices prior to HCV treatment may require surveillance endoscopy every 2-3 years, though the risk of a bleeding after SVR is generally low.168
The main change over time has been in the number of patients who are able to achieve SVR, particularly those with advanced disease. During the era of interferon-based therapy the treatment of patients with advanced liver disease/cirrhosis was more challenging. There were high rates of adverse events and, in some cases, a worsening of liver function on treatment. In addition, SVR rates were lower in those with advanced disease. Many of these patients can now achieve SVR, resulting in a larger number of patients with advanced disease remaining in long-term follow-up.
Cure research for HBV is very active and has been the subject of recent detailed reviews.169 There is a wide range of new potential treatments under investigation and significant interest from the pharmaceutical industry. Key barriers to curing HBV, which distinguish it from hepatitis C, include the ability of the virus to integrate into the DNA inside the patient’s cells, to establish small amounts of viral DNA inside cells which work as a “mini-chromosome” allowing persistence of infection even when effective antiviral is used (known as closed covalent circular DNA, cccDNA) and to weaken the body’s immune response to HBV in chronic infection.
There are different definitions of cure within hepatitis B research. Sterilising cure is the ultimate goal for drugs in development. Ideally, this would involve a finite treatment following completion of which there is no detectable virus (HBsAg and HBV DNA) in the patient’s serum, and eradication of both cccDNA and integrated HBV DNA from the body. However, this ambition remains a long way off with current technologies, so there is greater focus on “functional cure”. This would mean that after finite treatment there is not complete eradication of viral DNA from the liver; but there is no detectable virus in patient serum and no long-term consequences of infection (fibrosis/scarring and/or liver cancer).
A range of strategies is being explored to achieve HBV cure: preventing the entry of hepatitis B into liver cells, directly targeting cccDNA and integrated HBV DNA, preventing the production of new viruses in infected cells, preventing release of virus from infected cells, and making a stronger immune response against HBV.
Although antiviral treatments (such as tenofovir or entecavir) can suppress HBV replication, some ongoing production of new virus can infect other cells. With the identification of the receptor for hepatitis B on liver cells (sodium taurocholate co-transporting polypeptide, NTCP), there has been interest in blocking HBV entry to cells. The most advanced drug, bulevirtide (Myrcludex B), competes with HBV to enter liver cells and appears to have some activity in clinical studies when combined with interferon. Recent data suggests this drug may also have some effect in treatment of HDV (as HDV uses the same receptor as HBV).
Once inside the infected cell, HBV DNA is released into the cell nucleus. Here it can become integrated into the cell’s own DNA or be used to generate cccDNA. Both of these can then be a source for generation of new virus and are key targets for potential cures. New technologies of gene editing are being explored to breakdown cccDNA and integrated DNA using a range of methods (including the recently discovered naturally occurring CRISPR/Cas9 systems). Whilst very specific, one of the key challenges is delivering these treatments into cells. The HBx protein of the virus is key to maintaining cccDNA and this is also a target for drug development.
cccDNA and integrated DNA produce molecules of ribonucleic acid (RNA) that are the basis for new virus production. These RNA sequences can be specifically targeted by antisense RNAs or short inhibitory RNAs (siRNAs) to prevent them from making new viruses. There have been some encouraging results from new siRNA treatments (JNJ3989 and ARB-1467).
Development of drugs to prevent the assembly of new viruses within infected cells is one of the most active areas of research, aiming to supplement established treatments with drugs blocking the generation. Drugs in development fall into two groups (CpAMs and CAMs). A number of drugs have shown evidence of effect on HBV in early trials (Phase I/II) including JNJ-0440, JNJ-6379, ABI-H0731 and GLS4.
By blocking the release of new viral particles from infected cells, the ongoing chain of infection can be interrupted, potentially helping cure. A new class of drugs is being trialled called nucleic acid polymers (such as REP 2139) that have shown promise in early clinical studies.
In parallel to the strategies above that target the life of the virus within an infected cell, it is possible to strengthen the body’s immune response to remove infected cells. Approaches currently being evaluated include therapeutic vaccination. This involves generating specific immune response against part of the virus (antigens) in a similar way to preventative vaccination. In addition, new cell therapies are being investigated which engineer a patient’s own immune cells to target HBV-infected cells.
A complementary approach is based on the recognition that long-term HBV infection can lead to exhaustion of the body’s cells that fight HBV. New treatments which restore the function of exhausted cells (such as PD1 inhibitors) have proven very effective in treating some cancers and their role is being explored in HBV.
In the absence of a single, highly effective, curative treatment, there is a growing consensus that effective progress to cure will likely involve the combination of a number of treatments. Significant progress to a functional cure can be expected over the coming years, but the prospect of a sterilising cure remains a more distant prospect.
Hepatitis B (HBV) and hepatitis C (HCV) account for 96% of all deaths from viral hepatitis in the world. In recent years, the burden of viral hepatitis has been better recognised, accounting for more deaths each year than HIV or malaria, and a similar number to tuberculosis (TB). There has long been an effective vaccine against hepatitis B, estimated to have already prevented over 300 million new infections. Although there is no effective vaccine for hepatitis C on the horizon, the availability of shorter, less toxic and more effective drugs has help strengthen international ambitions to eliminate viral hepatitis. In 2016, the WHO developed targets and a strategy for the elimination of viral hepatitis as a public health threat by 2030.70 Although this falls short of eradication (the permanent reduction to zero of all infections), these are nonetheless ambitious goals. The target for mortality is a reduction of 10% due to viral hepatitis by 2020 and 65% by 2030, alongside a reduction in new infections (incidence) of 30% by 2020 and 90% by 2030. Achieving these goals will require substantial increases in access to both prevention and treatment for HBV and HCV. The Global Fund has raised major international funding to support national efforts towards control and elimination of HIV, TB and malaria. However, no such global mechanism yet exists for viral hepatitis. In its absence there will be reliance on national programmes, often in high burden/resource limited settings where health budgets are already stretched. Twenty countries account for over 80% of the burden of viral hepatitis globally, and each will need funded and effective elimination plans to achieve WHO targets.170
The WHO has identified key areas where progress is required to achieve goals and which will need to be part of national elimination plans.171 Prevention is critical. For hepatitis B, coverage of the effective vaccine has improved significantly (84% of those who need it have had 3 doses), but aims to reach 90%. A high proportion of new HBV infections in the world occur with transmission from mother to baby, and many can be prevented by birth dose vaccination. Support for delivery of birth dose vaccine has recently been announced by Gavi, the Vaccine Alliance.
Other means of preventing ongoing transmission of HBV and HCV are needed. To protect the general population there should be universal testing of blood products and although there has been great progress, coverage still falls short of 100% globally.172 Transmission through medical practice, particularly contaminated needles and syringes, remains an important driver of new infections and has been the subject of public health campaigns in Asia, notably Pakistan.
Ongoing transmission of hepatitis, particularly hepatitis C, is commonly associated with high-risk behaviour, particularly injecting drug use. Targets have been set for provision of clean needles and syringes. Criminalisation of injecting drug use remains a barrier to prevention in some high burden countries such as Russia.
Achieving the treatment targets (80% of all those in need of treatment to receive it by 2030) will require a substantial scale up in screening and testing for infection. It has been estimated that 90% of the 70 million individuals infected with HCV will need to be tested, in order for WHO targets to be met by 2030. This will require testing hundreds of millions of individuals. There is a need for better diagnostics that can be used for community screening and to make tests available at an affordable price.
Once linked to care, it is essential for treatment to be available to all who need it. Cost of drugs and the diagnostics required to use them remain a major issue in many high burden countries despite efforts to make treatments available from generic suppliers in many low-income settings.
Although these challenges are substantial, there are examples of low-income countries where major investments have been made. Perhaps the best example is Egypt, which has one of the largest epidemics of hepatitis C, as a result of contaminated needles used during a national schistosomiasis elimination programme. Egypt has developed locally produced generic treatments and committed to test over 50 million people in an effort to eliminate HCV by 2022.
The UK is one of a handful of wealthier nations currently on track to achieve WHO elimination targets, likely ahead of 2030. Although very high prices of new hepatitis C treatment meant limited access to those infected between 2015 and 2019, testing and treatment for both HBV and HCV is now widely available to all those who need it and HBV vaccine is now recommended routinely. Formal elimination plans have not been published, but WHO targets have been incorporated into public health planning.
The Public Health England strategy for infectious diseases (2020-25) has amongst its aims to “work with partners to provide surveillance data and facilitate test and treat strategies addressing hepatitis B and C;”173 meanwhile NHS England specialist commissioning has stated that it intends to achieve elimination of HCV by 2025, and has established a procurement deal with the pharmaceutical industry that incentivises increased diagnosis and treatment rates.
In 2008, the Scottish Government launched a hepatitis C Action Plan and in 2019 it published a hepatitis C Elimination Strategy—one that would ensure that Scotland met the WHO elimination targets by 2024. In alignment with the WHO targets, Scotland has its own burden of disease targets: a reduction in the prevalence of infection from 21,000 in 2018 to 5,000 by 2024; and a reduction in the annual numbers of hepatitis C-related deaths and new presentations of hepatitis C-related liver failure and hepatocellular carcinoma to single figures for each outcome. The ambition is for 3,000 infected people treated/year between 2020/21 and 2023/24.73
Northern Ireland is a low prevalence country for viral hepatitis (with approximately 100 cases of new infection of HBV and HCV respectively each year). The hepatitis B and C Managed Clinical Network plans to work with the Department of Health in Northern Ireland to produce an Action Plan towards the WHO elimination goals. In the meantime, the upscaling of the new and highly effective hepatitis C treatments and the introduction of hepatitis B vaccine for babies into the routine schedule means that this goal is moving a step closer in Northern Ireland.
A vaccine for hepatitis B has been available in the UK since 1982 and was introduced as part of the childhood vaccination programme in 2017 (see Q15.3). The vaccine is highly effective with low adverse event rates; as a result, there is relatively little development work in HBV vaccination. The first new HBV vaccine for 25 years was approved in 2017 and requires two, rather than three, doses to be effective.
Some groups of patients, including the elderly and those with immunosuppression, do not develop such strong immune responses and there is some interest in developing vaccine strategies that improve this.
In contrast, there is no vaccine available for hepatitis C and none likely to be available in the foreseeable future. Developing a hepatitis C vaccine is challenging for both biological and commercial reasons. The fact that approximately 20% of HCV infections clear infection spontaneously suggests that it is possible to generate immune responses that are protective against infection, although such individuals can often still be re-infected with HCV. The most important parts of the immune response protecting from HCV are not completely defined; both B cells and T cells seem to have a critical role in viral control.
Vaccine efforts focused on the development of B cell vaccines generating antibodies against the viral envelope (such as E1/E2 protein vaccine, HVR delta E2 protein) are challenged by the genetic diversity of the virus, though one has recently been tested in early clinical trials.174 Vaccines have been developed that produce strong T cell responses to HCV and are being evaluated (ChAd3/MVA-for example), though a recent early stage clinical trial (ClinicalTrials.gov Identifier NCT01436357) produced disappointing results.
Most development work for HCV vaccines is happening in academia, with little commercial interest. The decline in new infection seen with widespread access to newer HCV treatments is a challenge to further investment and will make efforts to test the efficacy of new vaccines harder.
For those living with HBV and HCV who develop advanced fibrosis/cirrhosis, fertility is reduced and pregnancy is uncommon.175 Women with cirrhosis may experience a loss of periods and increased rates of pregnancy loss, pre-term labour, foetal abnormality and perinatal death. In men with cirrhosis, low sperm counts and erectile dysfunction are common, and may be affected by medication (examples include spironolactone) taken for advanced liver disease (see Q15.11).176
Aside from those with advanced liver disease, there is more conflicting evidence of the effect of chronic HBV and HCV infection on fertility, and a full review of the evidence is beyond this report. Studies seeking to determine the effect of the virus per se need to account for liver disease and other confounding factors. This is not the case in many published studies which lack robust control groups to allow the effect of the virus itself to be teased out.
It should be noted that where a couple is trying to conceive and one partner is infected with HBV or HCV, the risks of transmission are very different (see Q15.5). HBV is more easily transmitted between couples, but can be largely prevented by effective vaccination of the negative partner. HCV, on the other hand, is uncommonly transmitted through vaginal intercourse, but there is no effective vaccine available (see supplemental responses).
In general, there is limited evidence that either HBV or HCV can have a direct effect on male fertility177 and no studies were identified that have looked at a general population of HBV positive women. In men, there is evidence that both HBV and HCV are associated with decreased sperm mobility.177-180 There is no evidence that either HBV or HCV has a direct effect on pregnancy, though both can be transmitted to neonates.
Aside from ribavirin and interferon, there is no evidence that current recommended treatments of hepatitis B or C affect fertility, and no studies were identified looking at sperm quality following DAA treatment.
Ribavirin is well recognised to be potentially teratogenic and it is advised not to become pregnant for six months after completion of therapy (either in the female or male partner), nor for it to be given in pregnancy.
Impotence is listed as a common side effect of interferon treatment (see Table 15.13a) and, although evidence is very limited, there are suggestions that interferon might lead to longer-term reductions in sperm count and motility.181
In vitro fertilisation (IVF) and embryo transfer are now routinely used to treat infertility associated with both male and female factors.
There is mixed evidence on whether maternal HBV infection can affect IVF outcomes. A large Chinese study of women undergoing in vitro fertilisation found a small increase in duration of infertility with more secondary infertility and ovulatory disorders in women with HBV. Embryo implantation rates were significantly lower in the HBeAG positive patients compared with the controls. However, the clinical pregnancy rate, miscarriage rate, live-birth rate, neonatal outcomes and pregnancy complications showed by the study were no different when comparing those with and without HBV.182
It has been shown that HBV can integrate into the genome of spermatozoa, raising the theoretical possibility of “diagonal transmission” where a HBV positive father may have an HBV positive child without the mother becoming infected.183 This is not a concern for HCV which does not integrate into the genome.
In general, where a female planning IVF is HCV positive, treatment should be completed prior to starting IVF. Whilst some studies have found an impact on implantation rate, abortion rate and pregnancy rates,184,185 many studies have found no association between success rates for IVF and HCV infection (whether of the male or female).180,186-188
Despite strict guidance to prevent healthcare-related transmission of viral hepatitis, including testing of potential parents, there have been rare reports of transmission of HCV to uninfected individuals during IVF (likely from contaminated medical equipment).189 In general, an IVF culture system should be a sterile system and there are clear guidelines to reduce the risk of transmission of the virus within the IVF procedures.183 However, transmission of HBV has been reported, for example as a result of a broken storage tank for eggs.190 Where samples known to be infected are stored, it has been recommended that they are kept separately from other samples.
Below is a summary of some of the key differences in burden, routes of transmission, disease progression, clinical presentation and treatment approaches for chronic HCV infection and chronic HBV infection in adolescents and children, compared to infection acquired in adulthood.
Most of the serious liver disease with HCV and HBV occurs in adults, and so most of the focus of the testing and treatment has been among adults. Much less attention has been paid to testing, treatment and prevention in children and adolescents. In part, this is because, until recently, none of the HCV DAA regimens had been approved for use in those younger than 18, and because of low rates of serious liver problems from HCV and HBV during childhood. Paediatricians and healthcare workers caring for children do not see the late complications of infection and tend not to test unless there is evidence of active hepatitis.
The greatest strides towards the elimination of HBV have been made with universal infant hepatitis B immunisation. This has been highly effective in reducing new infections in children in countries where this policy has been implemented. The UK only adopted this policy in 2017; the consequence of late adoption of universal immunisation is that the majority of UK-born and raised children and young adults are currently susceptible to infection with HBV.
Vertical and early childhood transmission are the main routes of HBV transmission globally (see also Q15.5), responsible for most chronic infections – including in adults who bear the greatest burden of morbidity and mortality. Universal birth dose and infant hepatitis B immunisation is the key preventative strategy for global elimination of HBV infection, and has been highly effective in reducing new vertical infections. Global progress on HBV testing and treatment, however, has been slow in adults and children. The estimated global prevalence in children aged 5 years or younger is 1.3%. Most children are in the “high replication, low level of inflammation” infection phase (see Q15.13) with normal or only minimally raised aminotransferases; cirrhosis and hepatocellular carcinoma are rare. Most adults have achieved a degree of immune control of HBV, indicating that at some point between infancy and adulthood, the human immune system ceases to tolerate HBV. There are significant gaps in understanding the triggers of an effective immune response during childhood or adolescence. Although entecavir is approved and recommended for children 2 to <18 years, and tenofovir for those 12 to <18 years, a conservative approach to treatment initiation is currently recommended, as a large proportion of children with high viral loads will naturally develop immune control.
The estimated global frequency and burden in children aged 1 to 18 years is 0·13% and 3.26 million (2.07-3.90) respectively. HCV infection is usually asymptomatic during childhood and serious liver diseases (cirrhosis and liver cancer) are rare. While more than eight different DAA combination treatments are available for adults, only two (sofosbuvir plus ribavirin and sofosbuvir/ledipasvir) have been approved for treatment in adolescents, and none are currently approved for children less than 12 years of age. Very few countries offer testing and treatment of adolescents and children as a public health intervention. As a result, globally only a fraction of HCV-infected children or adolescents have been diagnosed or treated, especially in poorer countries.
Children: The greatest strides towards achieving elimination of HBV have been made with universal infant hepatitis B immunisation. This has been highly effective in reducing new infections in children. In 2015, the WHO estimated the HBsAg prevalence globally in children under five years of age was about 1·3%, which corresponded to approximately 1·8 million (1·6–2·2) new infections in children globally.191
Adults: The main routes of acquiring new adult infections are through unsafe injections and sexual transmission among men who have sex with men or heterosexual persons with multiple sexual partners (see Q15.5).
Children: Mother to child transmission accounts for most infections in children in high prevalence countries (seroprevalence >8%) where infant vaccination (and especially birth dose) implementation has been suboptimal. Up to a third of new HBV infections may be due to horizontal early childhood transmission through child-to-child, household and intrafamilial transmission. Transmission can also be caused by poor infection control and lack of injection safety during medical, surgical and dental procedures or traditional practices (such as scarification or circumcision).
The age at infection determines the risk of developing chronic infection and progressive liver disease, and the development of complications such as cirrhosis and HCC mainly in adulthood. Hepatitis B infection acquired in adulthood is more often an acute, symptomatic but self-resolving infection, and uncommonly (less than 5%) leads to chronic infection. When hepatitis B infection is acquired at birth or in early childhood it is very likely to lead to chronic infection (more than 90%).
Most children are in the “high replication, low level of inflammation” immunotolerant phase of infection with normal or only minimally raised aminotransferases. Cirrhosis and hepatocellular carcinoma are rare with cirrhosis being reported in 1–5% of HBeAg-positive children. As noted above, at an unpredictable timepoint between infancy and adulthood the human immune system ceases to tolerate HBV. There are significant gaps in understanding the triggers and mechanisms of an effective immune response during childhood or adolescence.
The greatest burden of serious complications and deaths from hepatitis B are in adults who were infected at birth or in early childhood. In adults, the focus has been on reducing complications and mortality due to chronic liver disease, through scale-up of testing and long-term antiviral treatment with tenofovir or entecavir to suppress the virus (see Q15.13).
Adolescents and children: In contrast to adults, there is very limited data to guide recommendations on the optimal timing and indications for treatment in adolescents and children. Regardless of age, treatment is recommended for all persons with cirrhosis, as well as in those with active hepatitis, including HBeAg positive or negative with raised liver enzymes and high HBV DNA levels or with other evidence of liver inflammation or fibrosis. Although entecavir is approved and recommended for children 2 to <18 years, and tenofovir for those 12 to <18 years, given the need for potentially life-long treatment, with unknown safety, adherence and resistance risks, a conservative approach to treatment initiation is currently recommended. For children and young people aged 3 to <18 yrs, with elevated HBV DNA levels and raised transaminases (immune active phase), but without advanced liver disease, 48 weeks of once-weekly subcutaneous injections of PEG-IFN-alpha is indicated. If this fails to achieve an effective immune response, with control of HBV replication and normalised transaminases, treatment with antivirals may be indicated to prevent advancing liver fibrosis. The European Medicines Agency (EMA) recently approved the use of tenofovir alafenamide (TAF) for children aged 12 years and older and weighing >35 kg, which is likely to become the drug of choice in this age group through adolescence.
Children: Until recently, the disease burden in children and adolescents was poorly documented and understood. Early reports, based on high-risk hospital-based studies found high rates of infection (up to 20%) among adolescents and children treated in hospital for malignancy or kidney failure, needing dialysis or who had surgery. Based on a recent global review, the global estimate for 2018 for chronic HCV prevalence in children and adolescents (0-18 years) was 3.26 million (2.07-3.90) or 0.13% (0.3% prevalence in high income and 0.6% in low income countries).192
Adults: Injecting drug use accounts for most adult infections in high and middle income countries (as well as sexual transmission among men who have sex with men (MSM)). However, in low and middle income countries infection is most commonly associated with unsafe injection practices and procedures in health-care facilities with inadequate infection control practices, such as renal dialysis units.193
Children: Since routine blood-bank screening for HCV infection was introduced, mother to child vertical transmission has now become the main route of HCV infection in children in high and middle income countries. Vertical transmission occurs in up to 5% of babies born to infected mothers (and 10% if the mother is also co-infected with HIV). While vertical transmission mainly affects younger children,194 infection through injection drug use is more common in adolescents.
In low income countries, transmission in hospital and health clinics due to exposure to unsafe medical procedures also plays an important role in the spread of infection, especially among children having surgery or injections.
A recent study from the UK of 1,049 persons infected with HCV in childhood found that 53% were infected through injecting drug use in adolescence and 24% through receiving contaminated blood products.
Adults: With adult-acquired infection spontaneous clearance occurs in around 30% of cases (15–45%) within six months.195
Children: Following mother to child transmission, around 25 to 40% of children spontaneously clear/cure the infection in the first four years of life. Another 6%-12% may clear the virus before adulthood and the remainder develop a chronic infection that persists into adulthood.
Chronic HCV infection in children generally has a milder course than in adults, and some, but not all, studies have shown slower progression in young children compared to those infected in adulthood. The majority of children and adolescents will be asymptomatic, and severe disease, such as cirrhosis or liver cancer, is seen in only 2% of HCV-infected children. Some 80% of children followed in two studies for two to three decades had normal livers on biopsy. However, findings from a recent study from the UK shows that disease progression is much higher in those infected for more than 10 years. In this study cirrhosis developed in one-third of those infected in childhood after an average time of 33 years, regardless of when or how they were infected. In both adolescents and children other conditions, such as iron overload, obesity, cancer, HIV infection and hepatitis B infection, may also accelerate the development of liver disease.
Importantly, studies have shown an impact of HCV infection on overall quality of life, as well as psychosocial and cognitive function, even in HCV-infected children without any symptoms (compared to those without HCV infection).
In adolescents 12 to 17 years: For HCV-infected children aged 12 years or more, guidelines now recommend treatment in all (as for adults). To date, only two treatments (sofosbuvir plus ribavirin and recently sofosbuvir/ledipasvir) had been approved for treatment in adolescents or children aged 12-17 years-old or ≥35 kg. In adolescents receiving DAAs the cure rate was around 98% with good tolerability and no serious side effects. In addition, there were significant improvements in social functioning and school performance following cure. There are also ongoing clinical trials using other DAA regimens, in particular sofosbuvir/velpatasvir and glecaprevir/pibrentasvir for adolescents (12-17 years) and children down to 3 years.
In children younger than 12 years: Until recently the only approved treatment for children younger than 12 years was 24 to 48 weekly injections of PEG-interferon (P-IFN) α-2a or -2b with twice-daily oral ribavirin (RBV). These regimens are associated with significant side effects during treatment, including anaemia. This was a problem for children and young people with red cell disorders and increased their transfusion requirements. Uncommon, but potentially irreversible, after-therapy side effects have been described, including thyroid disease, type 1 diabetes, eye complications and growth impairment. For these reasons use of PEG-IFN / RBV treatment in those less than 12 years is currently limited to the few children with persistently abnormal liver blood tests, serious liver disease or with HIV co-infection. Until oral DAA regimens are approved for use in younger age groups, follow-up without treatment until adolescence has been preferred for most children. This is because of the generally low rate of serious liver disease in children less than 12 years; and the likely approval of additional DAAs within the next 12 months down to 3 years (Table 1).
In September 2019 sofosbuvir/ledipasvir was the first DAA regimen to be approved for use in children down to 3 years, but it is not a pan-genotypic regimen. The study showed a cure rate of 99% (89/90) and side effect events were mild. It is anticipated that other DAA treatments will be approved in children aged 3 to 11 years during 2020. Once DAAs are approved in children 3 to 12 years old, treatment with DAAs may then be considered for all children to eradicate the infection as early as possible, regardless of the stage of liver disease. This offers the opportunity for children to grow up free of the potential stigma and psychological consequences of a chronic infection.
With research now suggesting that disease progression begins even at a young age, early diagnosis and treatment can be key to preventing long-term morbidity. Several guidelines on viral hepatitis testing and treatment in children and adolescents recommend that children of infected mothers should be tested, as well as children where hepatitis is suspected because of symptoms or abnormal liver tests. Initial testing for HCV infection is recommended only after 18 months of age because spontaneous clearance/cure following mother to child transmission may continue to occur up to four years of age. Infection in those <18 months of age can only be confirmed by direct measurement of the HCV virus in the blood.
|
Guideline |
Recommendations for adolescents |
|
AASLD196 |
|
|
EASL197 |
|
|
WHO198 |
|
|
APASL199 |
|
|
ESPGHAN200 |
|
Table 21.a Comparison of the recommendations for treatment of chronic hepatitis C virus infection in children and adolescents from international guidelines – AASLD196, EASL197, WHO198, APASL199 and ESPGHAN.200 (Please note that APASL do not produce guidelines, but they have been included for the sake of completeness.)
This is addressed in response to question Q15.9.
The details of individual treatments and adverse events are outlined in Q15.13. With respect to HBV, most patients today will be prescribed tenofovir (TDF) as first line treatment. In discussions with patients as to the need for follow-up and monitoring, it would be expected to include consideration of potential toxicity to the kidneys, and the need for blood and urine monitoring.
In the rare circumstances of interferon use (for HBV rather than HCV in the current era), a discussion on the potential for effects on mood and cognitive function (as outlined in Q15.13) would be routine when considering the pros and cons of treatment. In particular, the potential impact on relationships and work would usually be discussed.
In the era of interferon-free treatment for HCV, the adverse events of treatment are now substantially less of a challenge than before and mental health would not routinely be a part of discussions. A short summary of potential adverse effects (in line with information available on the HCV Trust website) would be expected, in line with the most common side effects described (see section 15.13). Where ribavirin is used, there may be a more detailed discussion with regard to symptoms of anaemia in particular.
The general principle of infection control when treating all patients, including those with hepatitis, is one of ‘standard precautions’ for all patient care. Standard precautions are the minimum infection prevention practices that apply in any setting where health care is delivered, regardless of suspected or confirmed infection status of the patient. Standard precautions are designed to reduce the risk of transmission of blood-borne and other pathogens to healthcare workers and other patients from both recognised and unrecognised sources.
Standard precautions include:
A known hepatitis B or C positive patient can be admitted to the open ward, unless there is a high risk of bleeding and environmental contamination, when a side room may be advised to limit spread of infected material.
For the past few decades hepatitis B has been one of the most significant occupational infectious risks for health care providers, mainly via needlestick injury. In addition to standard precautions, immunisation against hepatitis B is one of the key factors in infection prevention for HCWs caring for patients with hepatitis B. In the UK pre-exposure hepatitis B immunisation has been recommended since the 1980s for all healthcare workers who may have direct contact with patients’ blood or body fluids, or staff who are at risk of injury from blood-contaminated sharp instruments or being deliberately injured or bitten by patients.202
There are currently no vaccines available against hepatitis C, although policies and procedures for the reporting of needlestick and splash injuries will ensure that a risk assessment takes place, and if exposed to HCV a diagnosis can be made early and treatment initiated. This would be expected to have a close to 100% rate of viral clearance.
The Health and Safety Executive (HSE) provides guidance203 for procedures for taking blood specimens and for clinical laboratories handling samples from patients with HBV or HCV in the course of their treatment. This includes the use of standard precautions for all patients, as all blood samples should be considered potentially infectious; therefore gloves should be worn when taking blood for all patients. When taking blood from individuals known or suspected to have a blood-borne virus infection, staff should also wear a protective apron. ‘Danger of infection’ labels should be used for samples known to pose a risk to staff handling the sample.
In 1972, the Rosenheim Advisory Group issued good practice guidelines to prevent the transmission of hepatitis B in renal dialysis and transplant units. Since then, with the identification of both HIV and HCV, new guidance on Good Practice Guidelines for Renal Dialysis/Transplantation Units (and the) Prevention and Control of Blood-borne Virus Infection has been published (2002), and remains current.204 Patients infected with HBV should ideally be dialysed in separate isolation facilities. Where these are not currently available, patients should be segregated in a separate area from other patients during dialysis. Patients infected with HCV should also be segregated from uninfected patients during dialysis. Because of the risk of cross infection, patients with different BBV infections should not be dialysed in a single segregated area at the same time.
Separate dedicated dialysis machines should be used for patients infected with HBV. Dedicated machines are not required for patients with HCV or HIV provided that cleaning and disinfection processes are properly carried out between patients according to the manufacturers’ instructions. Whenever possible, staff should nurse only infected or uninfected patients during a shift. If this is not practicable, more experienced staff should be assigned the task of caring for a mixed group of patients.
Whenever blood or body fluid is present, there is a potential risk of blood-borne transmission and appropriate protective measures should be taken. Only leakage of blood or body fluids produces a risk of BBV infection and simple hygiene measures are adequate to prevent transmission. However, whilst hygienic preparation is acceptable, current HSE guidance205 stipulates that BBV infected bodies can only be embalmed if additional transmission based precautions are in place, with appropriately robust measures for the use of sharps (minimise use or use safer sharps devices, for example) as this presents significant risk of exposure to workers.
REFERENCES
1. Kwo PY, Cohen SM, Lim JK. ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries. The American journal of gastroenterology 2017; 112(1): 18-35.
2. Stanaway JD, Flaxman AD, Naghavi M, et al. The global burden of viral hepatitis from 1990 to 2013: findings from the Global Burden of Disease Study 2013. Lancet 2016; 388(10049): 1081-8.
3. Tedder RS, Rodger AJ, Fries L, et al. The diversity and management of chronic hepatitis B virus infections in the United Kingdom: a wake-up call. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 2013; 56(7): 951-60.
4. Kao JH. Molecular epidemiology of hepatitis B virus. The Korean journal of internal medicine 2011; 26(3): 255-61.
5. Shi W, Zhang Z, Ling C, et al. Hepatitis B virus subgenotyping: history, effects of recombination, misclassifications, and corrections. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases 2013; 16: 355-61.
6. Hedskog C, Parhy B, Chang S, et al. Identification of 19 Novel Hepatitis C Virus Subtypes-Further Expanding HCV Classification. Open forum infectious diseases 2019; 6(3): ofz076.
7. Messina JP, Humphreys I, Flaxman A, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015; 61(1): 77-87.
8. Chen HY, Shen DT, Ji DZ, et al. Prevalence and burden of hepatitis D virus infection in the global population: a systematic review and meta-analysis. Gut 2018.
9. Wranke A, Pinheiro Borzacov LM, Parana R, et al. Clinical and virological heterogeneity of hepatitis delta in different regions world-wide: The Hepatitis Delta International Network (HDIN). Liver international: official journal of the International Association for the Study of the Liver 2018; 38(5): 842-50.
10. Blumberg BS, Alter HJ, Visnich S. A “New” Antigen in Leukemia Sera. Jama 1965; 191: 541-6.
11. Dane DS, Cameron CH, Briggs M. Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet 1970; 1(7649): 695-8.
12. Moulias RL, Couleru O, Goust JM. Viral hepatitis B and Dane particles. The New England journal of medicine 1973; 288(26): 1409.
13. Wrobel DM, Feinman V, Berris B, Sinclair J. Frequency of hepatitis B antigen in blood donors. Canadian Medical Association journal 1973; 108(5): 570-2.
14. Wallace J, Milne GR, Barr A. Total screening of blood donations for Australia (hepatitis associated) antigen and its antibody. British medical journal 1972; 1(5801): 663-4.
15. Connolly JH, McClelland WM, O’Neill HJ, Crowley D. Hepatitis B virus infection in Northern Ireland 1970-1987. The Ulster medical journal 1989; 58(1): 72-82.
16. Polakoff S. Public Health Laboratory Service surveillance of prophylaxis by specific hepatitis B immunoglobulin in England and Wales during the period 1975-1987. The Journal of infection 1990; 21(2): 213-20.
17. Ako TI, Follett EA, Dewar RD, Cossar JH, Reid D. Surveillance of hepatitis B virus infection in Scotland, 1973-1982. The Journal of hygiene 1984; 93(2): 233-6.
18. AGH. https://www.gov.uk/government/groups/advisory-group-on-hepatitis (accessed 24th December 2019).
19. Sullman SF, Zuckerman AJ. Viral hepatitis in drug addicts. Postgraduate medical journal 1971; 47(549): 473-5.
20. Polakoff S. Acute viral hepatitis B, reported to the Public Health Laboratory service. The Journal of infection 1990; 20(2): 163-8.
21. Polakoff S, Tillett HE. Acute viral hepatitis B: laboratory reports 1975-9. Br Med J (Clin Res Ed) 1982; 284(6332): 1881-2.
22. Archer AC, Cohen BJ, Mortimer PP. The value of screening blood donors for antibody to hepatitis B core antigen. Journal of clinical pathology 1983; 36(8): 924-8.
23. Krugman S. Hepatitis virus vaccines: present status. The Yale journal of biology and medicine 1982; 55(3-4): 375-81.
24. Gerlich WH. Medical virology of hepatitis B: how it began and where we are now. Virology journal 2013; 10: 239.
25. Goldberg D, McMenamin J. The United Kingdom’s hepatitis B immunisation strategy--where now? Communicable disease and public health 1998; 1(2): 79-83.
26. Glanz A, Byrne C, Jackson P. Role of community pharmacies in prevention of AIDS among injecting drug misusers: findings of a survey in England and Wales. Bmj 1989; 299(6707): 1076-9.
27. Stimson GV, Alldritt L, Dolan K, Donoghoe M. Syringe exchange schemes for drug users in England and Scotland. Br Med J (Clin Res Ed) 1988; 296(6638): 1717-9.
28. Choo QL, Weiner AJ, Overby LR, Kuo G, Houghton M, Bradley DW. Hepatitis C virus: the major causative agent of viral non-A, non-B hepatitis. British medical bulletin 1990; 46(2): 423-41.
29. Kuo G, Choo QL, Alter HJ, et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science 1989; 244(4902): 362-4.
30. Donahue JG, Munoz A, Ness PM, et al. The declining risk of post-transfusion hepatitis C virus infection. The New England journal of medicine 1992; 327(6): 369-73.
31. Chaudhary RK, MacLean C. Evaluation of first- and second-generation RIBA kits for detection of antibody to hepatitis C virus. Journal of clinical microbiology 1991; 29(10): 2329-30.
32. Hope RL, Weltman M, Dingley J, et al. Interferon alfa for chronic active hepatitis B. Long term follow-up of 62 patients: outcomes and predictors of response. The Medical journal of Australia 1995; 162(1): 8-11.
33. Poynard T, Bedossa P, Chevallier M, et al. A comparison of three interferon alfa-2b regimens for the long-term treatment of chronic non-A, non-B hepatitis. Multicenter Study Group. The New England journal of medicine 1995; 332(22): 1457-62.
34. Hoofnagle JH, Mullen KD, Jones DB, et al. Treatment of chronic non-A,non-B hepatitis with recombinant human alpha interferon. A preliminary report. The New England journal of medicine 1986; 315(25): 1575-8.
35. Hutchinson SJ, Goldberg DJ, King M, et al. Hepatitis C virus among childbearing women in Scotland: prevalence, deprivation, and diagnosis. Gut 2004; 53(4): 593-8.
36. Harris HE, Ramsay ME, Heptonstall J, Soldan K, Eldridge KP, Group HCVNRS. The HCV National Register: towards informing the natural history of hepatitis C infection in the UK. Journal of viral hepatitis 2000; 7(6): 420-7.
37. Ramsay ME, Balogun MA, Collins M, Balraj V. Laboratory surveillance of hepatitis C virus infection in England and Wales: 1992 to 1996. Communicable disease and public health 1998; 1(2): 89-94.
38. Shaw L, Taylor A, Roy KM, et al. Establishment of a database of diagnosed HCV-infected persons in Scotland. Communicable disease and public health 2003; 6(4): 305-10.
39. Taylor A, Goldberg D, Hutchinson S, Cameron S, Fox R. High risk injecting behaviour among injectors from Glasgow: cross sectional community wide surveys 1990-1999. Journal of epidemiology and community health 2001; 55(10): 766-7.
40. Weild AR, Gill ON, Bennett D, Livingstone SJ, Parry JV, Curran L. Prevalence of HIV, hepatitis B, and hepatitis C antibodies in prisoners in England and Wales: a national survey. Communicable disease and public health 2000; 3(2): 121-6.
41. Van Damme P, Kane M, Meheus A. Integration of hepatitis B vaccination into national immunisation programmes. Viral Hepatitis Prevention Board. Bmj 1997; 314(7086): 1033-6.
42. DOH. Hepatitis B antenatal screening and newborn immunisation programme; Best Practice guidance, 2011.
43. Hoofnagle JH, Lau D. New therapies for chronic hepatitis B. Journal of viral hepatitis 1997; 4 Suppl 1: 41-50.
44. Lai CL, Chien RN, Leung NW, et al. A one-year trial of lamivudine for chronic hepatitis B. Asia Hepatitis Lamivudine Study Group. The New England journal of medicine 1998; 339(2): 61-8.
45. Poynard T, Marcellin P, Lee SS, et al. Randomised trial of interferon alpha2b plus ribavirin for 48 weeks or for 24 weeks versus interferon alpha2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. International Hepatitis Interventional Therapy Group (IHIT). Lancet 1998; 352(9138): 1426-32.
46. McHutchison JG, Gordon SC, Schiff ER, et al. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. The New England journal of medicine 1998; 339(21): 1485-92.
47. Dusheiko G, Main J, Thomas H, et al. Ribavirin treatment for patients with chronic hepatitis C: results of a placebo-controlled study. Journal of hepatology 1996; 25(5): 591-8.
48. Roth WK, Weber M, Seifried E. Feasibility and efficacy of routine PCR screening of blood donations for hepatitis C virus, hepatitis B virus, and HIV-1 in a blood-bank setting. Lancet 1999; 353(9150): 359-63.
49. Cardoso MS, Koerner K, Kubanek B. Mini-pool screening by nucleic acid testing for hepatitis B virus, hepatitis C virus, and HIV: preliminary results. Transfusion 1998; 38(10): 905-7.
50. EASL. EASL Recommendations on Treatment of Hepatitis C 2018. Journal of hepatology 2018; 69(2): 461-511.
51. Hope VD, Ncube F, Hickman M, Judd A, Parry JV. Hepatitis B vaccine uptake among injecting drug users in England 1998 to 2004: is the prison vaccination programme driving recent improvements? Journal of viral hepatitis 2007; 14(9): 653-60.
52. Hutchinson SJ, Wadd S, Taylor A, et al. Sudden rise in uptake of hepatitis B vaccination among injecting drug users associated with a universal vaccine programme in prisons. Vaccine 2004; 23(2): 210-4.
53. Sutton AJ, Gay NJ, Edmunds WJ, et al. Modelling the hepatitis B vaccination programme in prisons. Epidemiology and infection 2006; 134(2): 231-42.
54. Trust H. History of the Hepatitis C Trust. : http://www.hepctrust.org.uk/about-us/history-hepatitis-c-trust (accessed 24th December 2019).
55. Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001; 358(9286): 958-65.
56. DOH. Hepatitis C Strategy for England, 2002.
57. DOH. Hepatitis C Action Plan for England, 2004.
58. WHA. World Hepatitis Alliance: Our Story. https://www.worldhepatitisalliance.org/about/our-story (accessed 24th December 2019).
59. DOH. Health clearance for tuberculosis, hepatitis B, hepatitis C and HIV: New healthcare workers, 2007.
60. Government S. Hepatitis C Action Plan for Scotland Phase II: May 2008-March 2011, 2008.
61. Government W. Blood Borne Viral Hepatitis Action Plan for Wales 2010-2015., 2010.
62. Kuhns MC, Busch MP. New strategies for blood donor screening for hepatitis B virus: nucleic acid testing versus immunoassay methods. Molecular diagnosis & therapy 2006; 10(2): 77-91.
63. Ward JW, Averhoff FM, Koh HK. World Hepatitis Day: a new era for hepatitis control. Lancet 2011; 378(9791): 552-3.
64. WHO. The Global Hepatitis Programme. 2011. https://www.who.int/hepatitis/about/global-hepatitis-programme/en/ (accessed 24th December 2019).
65. NICE. Hepatitis B and C testing: people at risk of infection Public Health Guideline [PH43]. 2012.
66. European Association For The Study Of The L. EASL clinical practice guidelines: Management of chronic hepatitis B virus infection. Journal of hepatology 2012; 57(1): 167-85.
67. Lawitz E, Gane EJ. Sofosbuvir for previously untreated chronic hepatitis C infection. The New England journal of medicine 2013; 369(7): 678-9.
68. WHO. Glasgow Declaration on Hepatitis, 2014.
69. Penrose Inquiry: Final Report, 2015.
70. WHO. Global Health Sector Strategy on Viral Hepatitis. Geneva: World Health Organization, 2016.
71. PHE. The hexavalent DTaP/IPV/Hib/HepB combination vaccine: information for healthcare practitioners about the inclusion of hepatitis B vaccine in the routine infant immunisation programme. 2017.
72. PHE. Hepatitis C in England 2019 : Working to eliminate hepatitis C as a major public health threat. 2019.
73. HPS. Scotland’s Hepatitis C Action Plan. 2019.
74. Shahnazarian V, Ramai D, Reddy M, Mohanty S. Hepatitis C virus genotype 3: clinical features, current and emerging viral inhibitors, future challenges. Annals of gastroenterology 2018; 31(5): 541-51.
75. Gupta N, Mbituyumuremyi A, Kabahizi J, et al. Treatment of chronic hepatitis C virus infection in Rwanda with ledipasvir-sofosbuvir (SHARED): a single-arm trial. The lancet Gastroenterology & hepatology 2019; 4(2): 119-26.
76. JPAC. Guidelines for the Blood Transfusion Services in the UK. 2013.
77. BSH. https://b-s-h.org.uk/guidelines/?category=Transfusion&p=1&search= (accessed 24th December 2019).
78. Blood Safety and Quality Regulations. 2005.
79. Safe Supplies: Monitor, inform, progress: NHS Blood and Transplant / Public Health England Epidemiology Unit, 2019.
80. SHOT. Annual Shot Report. 2018.
81. WHO. Guidelines on Estimation of Residual Risk of HIV, HBV or HCV Infections via Cellular Blood Components and Plasma. WHO Technical Report Series No 1004 2017.
82. Candotti D, Assennato SM, Laperche S, Allain JP, Levicnik-Stezinar S. Multiple HBV transfusion transmissions from undetected occult infections: revising the minimal infectious dose. Gut 2019; 68(2): 313-21.
83. Mavilia MG, Wu GY. Mechanisms and Prevention of Vertical Transmission in Chronic Viral Hepatitis. Journal of clinical and translational hepatology 2017; 5(2): 119-29.
84. Lin X, Guo Y, Zhou A, et al. Immunoprophylaxis failure against vertical transmission of hepatitis B virus in the Chinese population: a hospital-based study and a meta-analysis. The Pediatric infectious disease journal 2014; 33(9): 897-903.
85. Hurtado CW, Golden-Mason L, Brocato M, Krull M, Narkewicz MR, Rosen HR. Innate immune function in placenta and cord blood of hepatitis C--seropositive mother-infant dyads. PloS one 2010; 5(8): e12232.
86. Yao GB. Importance of perinatal versus horizontal transmission of hepatitis B virus infection in China. Gut 1996; 38 Suppl 2: S39-42.
87. Van Damme P, Cramm M, Van der Auwera JC, Vranckx R, Meheus A. Horizontal transmission of hepatitis B virus. Lancet 1995; 345(8941): 27-9.
88. Terrault NA, Dodge JL, Murphy EL, et al. Sexual transmission of hepatitis C virus among monogamous heterosexual couples: the HCV partners study. Hepatology 2013; 57(3): 881-9.
89. Blumberg BS, Gerstley BJ, Hungerford DA, London WT, Sutnick AI. A serum antigen (Australia antigen) in Down’s syndrome, leukemia, and hepatitis. Annals of internal medicine 1967; 66(5): 924-31.
90. England PH. Health Protection Report. 2016; 10(24).
91. Houghton M. The long and winding road leading to the identification of the hepatitis C virus. Journal of hepatology 2009; 51(5): 939-48.
92. Candotti D, Allain JP. Transfusion-transmitted hepatitis B virus infection. Journal of hepatology 2009; 51(4): 798-809.
93. Gretch DR. Diagnostic tests for hepatitis C. Hepatology 1997; 26(3 Suppl 1): 43S-7S.
94. Iriana S, Curry MP, Afdhal NH. Neurologic Manifestations of Hepatitis C Virus Infection. Clinics in liver disease 2017; 21(3): 535-42.
95. Yarlott L, Heald E, Forton D. Hepatitis C virus infection, and neurological and psychiatric disorders – A review. Journal of advanced research 2017; 8(2): 139-48.
96. EASL. Clinical Practice Guidelines on the management of hepatitis B virus infection. 2017.
97. Papatheodoridis GV, Chan HL, Hansen BE, Janssen HL, Lampertico P. Risk of hepatocellular carcinoma in chronic hepatitis B: assessment and modification with current antiviral therapy. Journal of hepatology 2015; 62(4): 956-67.
98. Marrero JA, Kulik LM, Sirlin CB, et al. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018; 68(2): 723-50.
99. Westbrook RH, Dusheiko G. Natural history of hepatitis C. Journal of hepatology 2014; 61(1 Suppl): S58-68.
100. Thein HH, Yi Q, Dore GJ, Krahn MD. Estimation of stage-specific fibrosis progression rates in chronic hepatitis C virus infection: a meta-analysis and meta-regression. Hepatology 2008; 48(2): 418-31.
101. Mahajan R, Xing J, Liu SJ, et al. Mortality among persons in care with hepatitis C virus infection: the Chronic Hepatitis Cohort Study (CHeCS), 2006-2010. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 2014; 58(8): 1055-61.
102. Wang T. Model of life expectancy of chronic hepatitis B carriers in an endemic region. Journal of epidemiology 2009; 19(6): 311-8.
103. Rose KM. Editorial commentary: Determining the effect of hepatitis C on mortality: sorting the signal from the noise. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 2014; 58(8): 1062-3.
104. Pinchoff J, Tran OC, Chen L, et al. Impact of hepatitis B on mortality and specific causes of death in adults with and without HIV co-infection in NYC, 2000-2011. Epidemiology and infection 2016; 144(16): 3354-64.
105. Pinchoff J, Drobnik A, Bornschlegel K, et al. Deaths among people with hepatitis C in New York City, 2000-2011. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 2014; 58(8): 1047-54.
106. Alavi M, Law MG, Grebely J, et al. Lower life expectancy among people with an HCV notification: a population-based linkage study. Journal of viral hepatitis 2014; 21(6): e10-8.
107. Mejia-Carvajal C, Czapek EE, Valentino LA. Life expectancy in hemophilia outcome. Journal of thrombosis and haemostasis : JTH 2006; 4(3): 507-9.
108. Plug I, Van Der Bom JG, Peters M, et al. Mortality and causes of death in patients with hemophilia, 1992-2001: a prospective cohort study. Journal of thrombosis and haemostasis : JTH 2006; 4(3): 510-6.
109. Mudge AM, Douglas C, Sansome X, et al. Risk of 12-month mortality among hospital inpatients using the surprise question and SPICT criteria: a prospective study. BMJ supportive & palliative care 2018; 8(2): 213-20.
110. Waziry R, Hajarizadeh B, Grebely J, et al. Hepatocellular carcinoma risk following direct-acting antiviral HCV therapy: A systematic review, meta-analyses, and meta-regression. Journal of hepatology 2017; 67(6): 1204-12.
111. Matsubara T, Sumazaki R, Shin K, Nagai Y, Takita H. Genotyping of hepatitis C virus: coinfection by multiple genotypes detected in children with chronic posttransfusion hepatitis C. Journal of pediatric gastroenterology and nutrition 1996; 22(1): 79-84.
112. Schijman A, Colina R, Mukomolov S, et al. Comparison of hepatitis C viral loads in patients with or without coinfection with different genotypes. Clinical and diagnostic laboratory immunology 2004; 11(2): 433-5.
113. Gomez-Mont Landerreche JG, Gil-Orbezo F, Morales-Dominguez H, Navarrete-Alvarez M, Trueba-Davalillo C, Capuano-Tripp P. [Nontraumatic causes of bilateral avascular necrosis of the femoral head: link between hepatitis C and pegylated interferon]. Acta ortopedica mexicana 2015; 29(3): 172-5.
114. Fletcher AM, Tellier P, Douville J, et al. Adverse vacuolation in multiple tissues in cynomolgus monkeys following repeat-dose administration of a PEGylated protein. Toxicology letters 2019; 317: 120-9.
115. Clark R. Drug review – Pegasys (peginterferon alfa-2a [40kDal]. Drugs Context 2017; 3(2): 65-83.
116. Webster R, Didier E, Harris P, et al. PEGylated proteins: evaluation of their safety in the absence of definitive metabolism studies. Drug metabolism and disposition: the biological fate of chemicals 2007; 35(1): 9-16.
117. Konopnicki D, Mocroft A, de Wit S, et al. Hepatitis B and HIV: prevalence, AIDS progression, response to highly active antiretroviral therapy and increased mortality in the EuroSIDA cohort. Aids 2005; 19(6): 593-601.
118. Thio CL, Seaberg EC, Skolasky R, Jr., et al. HIV-1, hepatitis B virus, and risk of liver-related mortality in the Multicenter Cohort Study (MACS). Lancet 2002; 360(9349): 1921-6.
119. Crowell TA, Gebo KA, Balagopal A, et al. Impact of hepatitis coinfection on hospitalization rates and causes in a multicenter cohort of persons living with HIV. Journal of acquired immune deficiency syndromes 2014; 65(4): 429-37.
120. Rockstroh JK, Spengler U, Sudhop T, et al. Immunosuppression may lead to progression of hepatitis C virus-associated liver disease in hemophiliacs coinfected with HIV. The American journal of gastroenterology 1996; 91(12): 2563-8.
121. Lo Re V, 3rd, Kallan MJ, Tate JP, et al. Hepatic decompensation in antiretroviral-treated patients co-infected with HIV and hepatitis C virus compared with hepatitis C virus-monoinfected patients: a cohort study. Annals of internal medicine 2014; 160(6): 369-79.
122. EACS. Guidelines V7.1. 2014.
123. Bischoff J, Mauss S, Cordes C, et al. Rates of sustained virological response 12 weeks after the scheduled end of direct-acting antiviral (DAA)-based hepatitis C virus (HCV) therapy from the National German HCV registry: does HIV coinfection impair the response to DAA combination therapy? HIV medicine 2018; 19(4): 299-307.
124. Mannucci PM, Gdovin S, Gringeri A, et al. Transmission of hepatitis A to patients with hemophilia by factor VIII concentrates treated with organic solvent and detergent to inactivate viruses. The Italian Collaborative Group. Annals of internal medicine 1994; 120(1): 1-7.
125. Huang YT, Yang HI, Liu J, Lee MH, Freeman JR, Chen CJ. Mediation Analysis of Hepatitis B and C in Relation to Hepatocellular Carcinoma Risk. Epidemiology 2016; 27(1): 14-20.
126. Liaw YF, Tsai SL, Sheen IS, et al. Clinical and virological course of chronic hepatitis B virus infection with hepatitis C and D virus markers. The American journal of gastroenterology 1998; 93(3): 354-9.
127. Hanley JP, Jarvis LM, Hayes PC, Lee AJ, Simmonds P, Ludlam CA. Patterns of hepatitis G viraemia and liver disease in haemophiliacs previously exposed to non-virus inactivated coagulation factor concentrates. Thrombosis and haemostasis 1998; 79(2): 291-5.
128. Gerolami V, Halfon P, Chambost H, et al. Prevalence of hepatitis G virus RNA in a monocentric population of French haemophiliacs. British journal of haematology 1997; 99(1): 209-14.
129. Schwarze-Zander C, Blackard JT, Rockstroh JK. Role of GB virus C in modulating HIV disease. Expert review of anti-infective therapy 2012; 10(5): 563-72.
130. Berg MG, Lee D, Coller K, et al. Discovery of a Novel Human Pegivirus in Blood Associated with Hepatitis C Virus Co-Infection. PLoS pathogens 2015; 11(12): e1005325.
131. Simmonds P, Manning A, Kenneil R, Carnie FW, Bell JE. Parenteral transmission of the novel human parvovirus PARV4. Emerging infectious diseases 2007; 13(9): 1386-8.
132. Lurcharchaiwong W, Chieochansin T, Payungporn S, Theamboonlers A, Poovorawan Y. Parvovirus 4 (PARV4) in serum of intravenous drug users and blood donors. Infection 2008; 36(5): 488-91.
133. Simmons R, Sharp C, McClure CP, et al. Parvovirus 4 infection and clinical outcome in high-risk populations. The Journal of infectious diseases 2012; 205(12): 1816-20.
134. Hsu TC, Chen TY, Lin MC, Tzang BS, Tsay GJ. Human parvovirus B19 infection in patients with chronic hepatitis B or hepatitis C infection. Journal of gastroenterology and hepatology 2005; 20(5): 733-8.
135. Caston JJ, Cantisan S, Gonzalez-Gasca F, et al. Interferon-gamma production by CMV-specific CD8+ T lymphocytes provides protection against cytomegalovirus reactivation in critically ill patients. Intensive care medicine 2016; 42(1): 46-53.
136. Abdelrahman T, Hughes J, Main J, McLauchlan J, Thursz M, Thomson E. Next-generation sequencing sheds light on the natural history of hepatitis C infection in patients who fail treatment. Hepatology 2015; 61(1): 88-97.
137. Darby SC, Ewart DW, Giangrande PL, et al. Mortality from liver cancer and liver disease in haemophilic men and boys in UK given blood products contaminated with hepatitis C. UK Haemophilia Centre Directors’ Organisation. Lancet 1997; 350(9089): 1425-31.
138. Seeff LB, Hollinger FB, Alter HJ, et al. Long-term mortality and morbidity of transfusion-associated non-A, non-B, and type C hepatitis: A National Heart, Lung, and Blood Institute collaborative study. Hepatology 2001; 33(2): 455-63.
139. Ludlam CA, Lee RJ, Prescott RJ, et al. Haemophilia care in central Scotland 1980-94. I. Demographic characteristics, hospital admissions and causes of death. Haemophilia : the official journal of the World Federation of Hemophilia 2000; 6(5): 494-503.
140. Federici AB, Santagostino E, Rumi MG, et al. The natural history of hepatitis C virus infection in Italian patients with von Willebrand’s disease: a cohort study. Haematologica 2006; 91(4): 503-8.
141. Marsella M, Ricchi P. Thalassemia and hepatocellular carcinoma: links and risks. Journal of blood medicine 2019; 10: 323-34.
142. Franchini M, Rossetti G, Tagliaferri A, et al. The natural history of chronic hepatitis C in a cohort of HIV-negative Italian patients with hereditary bleeding disorders. Blood 2001; 98(6): 1836-41.
143. Fujimura Y, Ishimoto S, Shimoyama T, et al. Genotypes and multiple infections with hepatitis C virus in patients with haemophilia A in Japan. Journal of viral hepatitis 1996; 3(2): 79-84.
144. Papadopoulos N, Argiana V, Deutsch M. Hepatitis C infection in patients with hereditary bleeding disorders: epidemiology, natural history, and management. Annals of gastroenterology 2018; 31(1): 35-41.
145. Hanley JP, Jarvis LM, Andrew J, et al. Interferon treatment for chronic hepatitis C infection in hemophiliacs--influence of virus load, genotype, and liver pathology on response. Blood 1996; 87(5): 1704-9.
146. Hezode C, Colombo M, Bourliere M, et al. Elbasvir/Grazoprevir for Patients With Hepatitis C Virus Infection and Inherited Blood Disorders: A Phase III Study. Hepatology 2017; 66(3): 736-45.
147. Uemura H, Tsukada K, Mizushima D, et al. Interferon-free therapy with direct acting antivirals for HCV/HIV-1 co-infected Japanese patients with inherited bleeding disorders. PloS one 2017; 12(10): e0186255.
148. Murthy V, Murray D, Hebballi S, et al. Outcome of liver transplantation in patients with hereditary bleeding disorders: a single centre UK experience. Haemophilia : the official journal of the World Federation of Hemophilia 2016; 22(3): e139-44.
149. Younossi ZM, Stepanova M, Charlton M, et al. Patient-reported outcomes with sofosbuvir and velpatasvir with or without ribavirin for hepatitis C virus-related decompensated cirrhosis: an exploratory analysis from the randomised, open-label ASTRAL-4 phase 3 trial. The lancet Gastroenterology & hepatology 2016; 1(2): 122-32.
150. Simmons B, Saleem J, Heath K, Cooke GS, Hill A. Long-Term Treatment Outcomes of Patients Infected With Hepatitis C Virus: A Systematic Review and Meta-analysis of the Survival Benefit of Achieving a Sustained Virological Response. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 2015; 61(5): 730-40.
151. Jakobsen JC, Nielsen EE, Koretz RL, Gluud C. Do direct acting antivirals cure chronic hepatitis C? Bmj 2018; 361: k1382.
152. Jakobsen JC, Nielsen EE, Feinberg J, et al. Direct-acting antivirals for chronic hepatitis C. The Cochrane database of systematic reviews 2017; 9: CD012143.
153. Cacoub P, Saadoun D, Bourliere M, et al. Hepatitis B virus genotypes and extrahepatic manifestations. Journal of hepatology 2005; 43(5): 764-70.
154. Han SH. Extrahepatic manifestations of chronic hepatitis B. Clinics in liver disease 2004; 8(2): 403-18.
155. Cacoub P, Gragnani L, Comarmond C, Zignego AL. Extrahepatic manifestations of chronic hepatitis C virus infection. Digestive and liver disease : official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver 2014; 46 Suppl 5: S165-73.
156. Mahale P, Sturgis EM, Tweardy DJ, Ariza-Heredia EJ, Torres HA. Association Between Hepatitis C Virus and Head and Neck Cancers. Journal of the National Cancer Institute 2016; 108(8).
157. Borman M, Swain MG. Hepatitis C virus treatment complicated by rheumatoid arthritis. Gastroenterology & hepatology 2011; 7(11): 774-6.
158. Kozanoglu E, Canataroglu A, Abayli B, Colakoglu S, Goncu K. Fibromyalgia syndrome in patients with hepatitis C infection. Rheumatology international 2003; 23(5): 248-51.
159. Dillon JF, Miller MH, Robinson EM, et al. Intelligent liver function testing (iLFT): A trial of automated diagnosis and staging of liver disease in primary care. Journal of hepatology 2019; 71(4): 699-706.
160. BASHH. CEG guidance on tests for Sexually Transmitted Infections. 2015.
161. Kessels RP. Patients’ memory for medical information. Journal of the Royal Society of Medicine 2003; 96(5): 219-22.
162. Elwyn G, Barr PJ, Grande SW. Patients recording clinical encounters: a path to empowerment? Assessment by mixed methods. BMJ open 2015; 5(8): e008566.
163. What Realistic Medicine Is and what it isn’t. https://www.realisticmedicine.scot/about/ (accessed 16th January 2020)
164. NHSE. Shared Decision Making. https://www.england.nhs.uk/shared-decision-making/ (accessed 16th January 2020)
165. Health, Eligibility and Travel. https://my.blood.co.uk/knowledgebase/Index/H (accessed 28th December 2019).
166. https://www.blood.co.uk/the-donation-process/further-information/tests-we-carry-out/ (accessed 28th December 2019).
167. AASLD-IDSA. Hepatitis C Guidance 2018 Update: AASLD-IDSA Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 2018; 67(10): 1477-92.
168. Dusheiko G. Hepatitis C in the EU: setting the terms for elimination. The lancet Gastroenterology & hepatology 2017; 2(5): 314-5.
169. Fanning GC, Zoulim F, Hou J, Bertoletti A. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nature reviews Drug discovery 2019; 18(11): 827-44.
170. Cooke GS, Andrieux-Meyer I, Applegate TL, et al. Accelerating the elimination of viral hepatitis: a Lancet Gastroenterology & Hepatology Commission. The lancet Gastroenterology & hepatology 2019; 4(2): 135-84.
171. WHO. Global Hepatitis Report, 2017. Geneva, Switzerland: World Health Organization, 2017.
172. WHO. Global status report on blood safety and availability 2016. Geneva, Switzerland: World Health Organization, 2017.
173. PHE. PHE Infectious Diseases Strategy 2020-25, 2019.
174. Law JL, Chen C, Wong J, et al. A hepatitis C virus (HCV) vaccine comprising envelope glycoproteins gpE1/gpE2 derived from a single isolate elicits broad cross-genotype neutralizing antibodies in humans. PloS one 2013; 8(3): e59776.
175. Westbrook RH, Dusheiko G, Williamson C. Pregnancy and liver disease. Journal of hepatology 2016; 64(4): 933-45.
176. Foresta C, Schipilliti M, Ciarleglio FA, Lenzi A, D’Amico D. Male hypogonadism in cirrhosis and after liver transplantation. Journal of endocrinological investigation 2008; 31(5): 470-8.
177. van Leeuwen E. The Impact of HIV, HBV and HCV on male and female fertility Assisted Reproductive Technologies and Infectious DIseases: Springer; 2016.
178. Garolla A, Pizzol D, Bertoldo A, Menegazzo M, Barzon L, Foresta C. Sperm viral infection and male infertility: focus on HBV, HCV, HIV, HPV, HSV, HCMV, and AAV. Journal of reproductive immunology 2013; 100(1): 20-9.
179. Hofny ER, Ali ME, Taha EA, et al. Semen and hormonal parameters in men with chronic hepatitis C infection. Fertility and sterility 2011; 95(8): 2557-9.
180. Safarinejad MR, Kolahi AA, Iravani S. Evaluation of semen variables, sperm chromosomal abnormalities and reproductive endocrine profile in patients with chronic hepatitis C. BJU international 2010; 105(1): 79-86.
181. Bukhari SA, Ahmed MM, Anjum F, et al. Post interferon therapy decreases male fertility through gonadotoxic effect. Pakistan journal of pharmaceutical sciences 2018; 31(4(Supplementary)): 1565-70.
182. Wang L, Li L, Huang C, et al. Maternal chronic hepatitis B virus infection does not affect pregnancy outcomes in infertile patients receiving first in vitro fertilization treatment. Fertility and sterility 2019; 112(2): 250-7 e1.
183. Steyaert SR, Leroux-Roels GG, Dhont M. Infections in IVF: review and guidelines. Human reproduction update 2000; 6(5): 432-41.
184. Hanafi NF, Abo Ali AH, Abo el kheir HF. ICSI outcome in women who have positive PCR result for hepatitis C virus. Human reproduction 2011; 26(1): 143-7.
185. Pirwany IR, Phillips S, Kelly S, Buckett W, Tan SL. Reproductive performance of couples discordant for hepatitis B and C following IVF treatment. Journal of assisted reproduction and genetics 2004; 21(5): 157-61.
186. Yang L, Zhao R, Zheng Y, Song X. Effect of hepatitis C virus infection on the outcomes of in vitro fertilization. International journal of clinical and experimental medicine 2015; 8(4): 6230-5.
187. Prisant N, Tubiana R, Lefebvre G, et al. HIV-1 or hepatitis C chronic infection in serodiscordant infertile couples has no impact on infertility treatment outcome. Fertility and sterility 2010; 93(3): 1020-3.
188. Molina I, Carmen Del Gonzalvo M, Clavero A, et al. Assisted reproductive technology and obstetric outcome in couples when the male partner has a chronic viral disease. International journal of fertility & sterility 2014; 7(4): 291-300.
189. Lesourd F, Izopet J, Mervan C, et al. Transmissions of hepatitis C virus during the ancillary procedures for assisted conception. Human reproduction 2000; 15(5): 1083-5.
190. Tedder RS, Zuckerman MA, Goldstone AH, et al. Hepatitis B transmission from contaminated cryopreservation tank. Lancet 1995; 346(8968): 137-40.
191. Indolfi G, Easterbrook P, Dusheiko G, et al. Hepatitis B virus infection in children and adolescents. The lancet Gastroenterology & hepatology 2019; 4(6): 466-76.
192. Indolfi G, Easterbrook P, Dusheiko G, et al. Hepatitis C virus infection in children and adolescents. The lancet Gastroenterology & hepatology 2019; 4(6): 477-87.
193. WHO. Global Hepatitis Report 2017. Geneva; Switzerland: World Health Organization, 2017.
194. Pawlowska M, Domagalski K, Pniewska A, Smok B, Halota W, Tretyn A. What’s new in hepatitis C virus infections in children? World J Gastroenterol 2015; 21(38): 10783-9.
195. Grebely J, Page K, Sacks-Davis R, et al. The effects of female sex, viral genotype, and IL28B genotype on spontaneous clearance of acute hepatitis C virus infection. Hepatology 2014; 59(1): 109-20.
196. AASLD-IDSA. Recommendations for testing, managing, and treating hepatitis C. https://www.hcvguidelines.org/ (accessed 22nd January 2020)
197. European Association for the Study of the Liver. EASL Recommendations on Treatment of Hepatitis C 2018. Journal of hepatology 2018; 69(2): 461-511.
198. WHO. Guidelines for the care and treatment of persons diagnosed with chronic hepatitis C virus infection. Geneva, Switzerland, 2018.
199. Omata M, Kanda T, Wei L, et al. APASL consensus statements and recommendation on treatment of hepatitis C. Hepatol Int. United States; 2016: 702-26.
200. Indolfi G, Hierro L, Dezsofi A, et al. Treatment of Chronic Hepatitis C Virus Infection in Children: A Position Paper by the Hepatology Committee of European Society of Paediatric Gastroenterology, Hepatology and Nutrition. Journal of pediatric gastroenterology and nutrition 2018; 66(3): 505-15.
201. HSE. Laundry treatments at high and low temperatures.
202. PHE. Hepatitis B immunisation information for public health professionals. The Green Book; 2013.
203. HSE. Safe working and the prevention of infection in clinical laboratories and similar facilities. 2003.
204. DOH. Good Practice Guidelines for Renal Dialysis/Transplantation Units. 2002.
205. HSE. Managing Infection Risks when Handling the Deceased. 2018.
This addendum answers the questions in the Further Supplemental Letter of Instruction. The numbering below follows the paragraph numbering in that Letter. These questions were raised during the expert hearings.
There are studies of the prevalence of different HCV genotypes in patients who received blood products in the UK. In general, the genotypes represent those of the population donating blood products (usually from the UK, northern Europe or USA). A published series1 of 302 individuals with haemophilia infected with hepatitis C and managed in Scotland found 67.4% to have genotype 1, 5.7% genotype 2, 25.4% genotype 3 and 1% genotype 4.
In an earlier study, also from Scotland2, the genotype distribution (G1 60%, G2 6.3%, G3 32%, G5 1.5%) was similar to that of Scottish blood donors. The study was notable as it looked for evidence that individuals may be infected with more than one genotype. The authors compared the genotypes found in the same patients across three periods of time (i) pre-1984 (chosen to be before the arrival of HIV) (ii) 1986-8 (heat treated factor VIII/IX used after 1985) and (iii) 1990-91. While most patients had the same genotype over time, nine (31%) changed genotype, suggesting infection with multiple genotypes. Where genotype changes were found, they suggest a trend to genotype 1 becoming more dominant over time.
A study from a cohort of 305 individuals with inherited coagulation disorders from a single London clinic found similar genotype distributions of genotypes, though with a slightly higher proportion of G2 than might have been expected from background populations (G1 63%, G2 14%, G3 18%)3. Variation in genotype distributions between cohorts will be affected by the makeup of donor populations and variation in the nature and type of blood products used at particular clinics.
The hepatitis B virus was discovered in 1964. Only a few years later studies on the prevalence of hepatitis B surface antibody (HBsAb) in populations with different numbers of sexual partners, including commercial sex workers, suggested the possibility of sexual transmission of hepatitis B surface antigen (HBsAg)4-6. So, by the early 1970s, the possibility of sexual transmission of hepatitis became increasingly evident.
REFERENCES
1. Khan MM, Tait RC, Kerr R, et al. Hepatitis C infection and outcomes in the Scottish haemophilia population. Haemophilia: The Official Journal of the World Federation of Hemophilia 2013; 19(6): 870-5.
2. Jarvis LM, Watson HG, McOmish F, Peutherer JF, Ludlam CA, Simmonds P. Frequent reinfection and reactivation of hepatitis C virus genotypes in multitransfused hemophiliacs. The Journal of Infectious Diseases 1994; 170(4): 1018-22.
3. Yee TT, Griffioen A, Sabin CA, Dusheiko G, Lee CA. The natural history of HCV in a cohort of haemophilic patients infected between 1961 and 1985. Gut 2000; 47(6): 845-51.
4. Papaevangelou G, Trichopoulos D, Kremastinou T, Papoutsakis G. Prevalence of hepatitis B antigen and antibody in prostitutes. British Medical Journal 1974; 2(5913): 256-8.
5. Heathcote J, Sherlock S. Spread of acute type-B hepatitis in London. The Lancet 1973; 1(7818): 1468-70.
6. Hersh T, Melnick JL, Goyal RK, Hollinger FB. Nonparenteral transmission of viral hepatitis type B (Australia antigen-associated serum hepatitis). The New England Journal of Medicine 1971; 285(24): 1363-4.
Each contributing group member confirms that he or she understands his or her duty to provide independent evidence and has complied with that duty.
All contributing group members confirm that in respect of those parts of the report to which they have contributed:
Professor Jane Anderson is chair of the National AIDS Trust and a past chair of the British HIV Association. She is a consultant physician in HIV medicine at Homerton University Hospital NHS Foundation Trust. She chairs the Public Health England Advisory Group for HIV and Sexual/Reproductive Health and she represents London clinicians in the NHS England Clinical Reference Group for HIV. She has worked as a clinician and researcher in HIV medicine since the virus emerged in the 1980s. She holds honorary academic appointments at Barts, The London School of Medicine and Dentistry and University College London. Her work focuses on ethnic minority and migrant populations in relation to HIV in the UK, with a particular focus on HIV care for women and families. Her wide-ranging work engages with the current medical, social, ethical and legal challenges posed by HIV. She is also a visiting fellow at The King’s Fund, an independent charity working to improve health and care in England.
Professor Graham Cooke is a National Institute for Health Research (NIHR) Research Professor of Infectious Diseases at Imperial College London, Honorary Consultant and Lead for co-infection services within Imperial College NHS Trust. Previously, he was based at the Africa Health Research Institute in KwaZulu-Natal. He led the Commission on Accelerating the Elimination of Viral Hepatitis published in 2019, chairs the WHO Committee on the Selection and Use of Essential Medicines and is a member of the National Viral Hepatitis Strategy Group. He chairs the British HIV Association (BHIVA) Hepatitis Expert Advisory Group which is leading efforts for the microelimination of hepatitis C in those living with HIV. His current work focuses on precision medicine for managing infectious diseases and access to medicines, particularly for HIV/viral hepatitis. He led the clinical workstream for the MRC Stratified Medicine Consortium (STOPHCV) from 2003-19, and is infection lead for the London In-vitro diagnostics cooperative. He was chief investigator on the STOPHCV-1 trial and currently has studies running in the UK and Vietnam, in collaboration with the MRC Clinical Trials Unit.
Professor John Dillon is a professor of Hepatology and Gastroenterology and a principle investigator in the Division of Molecular and Clinical Medicine, at the University of Dundee, based at Ninewells Hospital, Dundee. He is also an honorary consultant with NHS Tayside, leading a busy general hepatology service and a research group. His research interests include; new pathways of care for patients with abnormal liver function tests and for people infected with HCV, new therapies for HCV infection, as well as novel diagnostics and treatments for non-alcoholic fatty liver disease. He has published over 150 peer reviewed original research papers. He chairs the Scottish HCV Action Plan Clinical Leads Group, is a member of the Scottish government’s ministerial advisory board for blood-borne viruses and sexual health and is the president of the Scottish Society of Gastroenterology. He previously led the development group of the UK consensus guidelines for HCV and has chaired both the Hepatitis C SIGN guideline development group, and the Scottish Health Action on Alcohol Problems (SHAAP) group that produced the recent “Alcohol-related liver disease: guidance for good practice” documents.
Professor Philippa Easterbrook is a senior scientist in the Global Hepatitis Programme, HIV department at the WHO Headquarters in Geneva. She is an HIV and infectious diseases physician and epidemiologist who has worked in the UK, United States and Sub-Saharan Africa. At the WHO, she led the development and dissemination of global normative guidance for HIV as well as hepatitis B and C testing and treatment. She also provides technical guidance to national programmes worldwide on the implementation of hepatitis B and C testing and treatment scale-up programmes as part of a global elimination strategy. For eleven years, she was head of department, professor of HIV Medicine, and consultant physician in Infectious Diseases at King’s College London, and also head of research at the Infectious Diseases Institute in Uganda. Professor Easterbrook has served as a member of the UK Medical Research Council Infection and Immunity Committee, and was vice-chair of the World Health Organization Guidelines Review Committee. Her HIV research has encompassed epidemiology, clinical trials, operational and qualitative research, and laboratory-based studies.
Professor David Goldberg is a consultant in Public Health Medicine and Clinical Epidemiology at Health Protection Scotland (HPS) who, over the last 25 years, has developed, implemented and evaluated interventions and monitoring techniques to prevent HIV and hepatitis B and C infections and their diseases, both nationally and internationally. He led the team which developed and coordinated the implementation of Scotland’s Hepatitis C Action Plan. Previous roles include Henry Mechan Professor of Public Health, Deputy Director of Health Protection; and former Acting Director of the Scottish Centre for Infection and Environmental Health. He is an honorary professor of Public Health at the University of Glasgow and a professor of Public Health at Glasgow Caledonian University. He serves on several Scottish committees and is involved in the postgraduate supervision and teaching of students affiliated to the University of Glasgow, and is the author of approximately 250 peer-reviewed articles. He currently chairs Scotland’s Hepatitis C Treatment and Therapies Group.
Dr Katie Hands is a Consultant Haematologist with the Scottish National Blood Transfusion Service (SNBTS) and is based at Ninewells Hospital in Dundee. She studied medicine at the University of Dundee, and during haematology specialty training undertook a PhD under the supervision of Professor Ron Hay. Dr Hands was appointed as a Consultant with SNBTS in 2016, providing transfusion medicine support for NHS Tayside, where her role includes promoting the safe and appropriate use of blood in all hospital departments. Her wider roles within SNBTS include policy development and transfusion medicine teaching as part of a National teaching programme for haematology registrars. She is a member of the British Society for Haematology Transfusion Task Force, and is involved in the preparation of evidenced based guidelines relating to all aspects of blood transfusion in the UK.
Dr Scott Jamieson is a General Practitioner in Kirriemuir, Scotland. He sits on the Royal College of General Practitioners (RCGP) Scottish Council, where he is the Executive Officer for Quality Improvement. He is Clinical Prescribing Lead for Angus Health and Social Care Partnership, sitting on the local Drug and Therapeutics Committee and the Non-Medicines Advisory Group. In addition to this, he is a GP trainer and GP representative on the Scottish Intercollegiate Guidelines Network (SIGN) Council, an organisation dedicated to improving the quality of health care for patients in Scotland by reducing variation in practice and outcome, through development and dissemination of national clinical guidelines. His interests include dermatology, minor surgery, and sexual and reproductive health.
Dr Katie Jeffery is a consultant in Clinical Infection, the Infection Control Doctor (ICD) and Director of Infection Prevention and Control (DIPC) at the Oxford University Hospitals (OUH) NHS Foundation Trust. She trained in Microbiology with a particular interest in Virology, and has a PhD in HTLV-1 infection. Her clinical interests are infection prevention and control, viral diagnostics, viral hepatitis, and infections in the immunocompromised host. Previously she was Clinical Lead for Microbiology in the OUH, where she led on the introduction of a number of new laboratory assays in serology and molecular diagnostics. She holds a number of positions of responsibility both locally and nationally, including being a Virology Examiner for the Royal College of Pathologists, the Vice President of the British Infection Association, and a member of the Expert Advisory Group for Infectious Diseases for the MHRA (Medicines and Healthcare products Regulatory Agency). She has nearly 20 years’ experience in treating patients with viral hepatitis, and is regularly involved as an investigator in national and international studies, especially those that benefit NHS patients by allowing early access to new drugs for the treatment of hepatitis C.
Dr David J Johnston OBE is a General Practitioner at Maine Medical Practice in County Antrim. He is a Clinical Director of Dalriada Urgent Care, an out of hours primary care provider for the north east of Northern Ireland . He is also a member of “Practice 400” which provides care for General Medical Service designated “violent patients” and was involved in the establishment of ECHO, a scheme to enhance primary care services for homeless patients. His interest areas include pre-hospital immediate medical care, out of hours primary care and rural medicine. He has been a GlaxoWelcome research fellow with the University of Ulster and was involved in researching rural General Practice in Northern Ireland. David served as chairman of the NI Council of the Royal College of General Practitioners (RCGP) from 2008 to 2011 and was also a member of the UK council of RCGP. He continues to chair a number of community charities.
Dr Aileen Marshall is a Consultant Hepatologist and Honorary Senior Lecturer at the Royal Free Hospital and UCL Institute of Liver and Digestive Health. Her interest areas include liver transplant medicine, hepatocellular carcinoma, autoimmune liver disease, and palliative care for patients with advanced liver disease. Dr Marshall is the service lead for Hepatology at the Royal Free NHS Foundation Trust, and Hepatology lead for the North Thames Clinical Research Network. She is also the chair of Hepatocellular Carcinoma UK, a multidisciplinary organisation affiliated to the British Association for the Study of Liver Diseases aimed at promoting collaborative basic and clinical research and improving access to treatment and quality of care for those suffering with the disease. She is a member of both the British Association for the Study of Liver Disease and the Royal College of Physician. Dr Marshall holds a PhD from the University of Cambridge in Clinical Medicine and obtained her CCT in Gastroenterology & General Medicine, subspecialising in Hepatology.
Professor Jürgen Rockstroh is Professor of Medicine and Head of the HIV Outpatient Clinic at the University of Bonn, Germany, which treats the world’s largest cohort of HIV-infected haemophiliacs. In addition to his clinical practice, Dr Rockstroh is involved in HIV research on: antiviral therapy, including new drug classes; the course of HIV disease in haemophiliacs; and HIV and hepatitis co-infection. He has been an investigator in multiple clinical trials of antiretroviral agents and treatments for HIV and hepatitis co-infection. He was the president of the German AIDS Society from 2007 to 2011, has been an executive committee member of the European AIDS Clinical Society (EACS) since 2009 and in 2019 was elected as president of EACS. Dr Rockstroh has been a member of the governing council of the International AIDS Society since 2011, and currently chairs the hepatitis research activities in NEAT (European AIDS treatment Network) and EuroSIDA. Between 2011 and 2017 he chaired the National German AIDS Advisory Panel, and the EACS co-infection guidelines. Dr Rockstroh has authored and co-authored over 500 publications in peer-reviewed journals, and over 70 book chapters. The German Society for Infectious Diseases awarded Dr Rockstroh the national AIDS research prize in 2005.
Dr Mallika Sekhar is a consultant haematologist and honorary senior lecturer at UCL. She specialises in myeloproliferative diseases and blood transfusion, across the University College London Hospital and Royal Free Hospitals, with a special interest in patients with vascular thrombosis and myeloproliferative diseases. She has been involved with writing Management Process Description (MPD) guidelines for the British Committee for the Standards in Haematology (BCSH). Dr Sekhar has been the Lead Investigator in studies on abdominal vein thrombosis in myeloproliferative diseases and transfusion in haematological malignancies, and a member of the National Cancer Research Institute (NCRI) MPD and supportive care clinical studies group. She was a member of the clinical expert panel on the Pathology Modernisation initiative for London, and is the lead for undergraduate education in haematology at the Royal Free campus. Previously, she was chair of the London Regional Council of the Royal College of Pathologists and has been a member of the National Blood Transfusion Committee Group on Education since 2012.
Professor Mark Thursz is head of the Department of Metabolism, Digestion and Reproduction, and professor of Hepatology at Imperial College London. He is director of the Imperial Biomedical Research Centre and an Honorary Consultant in Gastroenterology and Hepatology at Imperial College Healthcare NHS Trust. He is a board member of the St Mary’s Development Trust, a member of the Imperial College Healthcare Charity Grants Committee, and a WHO Global Hepatitis Team advisor. He is the previous chief investigator of PROLIFICA and co-designed the first PROLIFICA studies on hepatitis B in West Africa, identifying strategies to reduce the burden of cirrhosis and liver cancer in Africa by controlling viral hepatitis infection. He is currently chief investigator on two multi-centre trials; the warfarin anticoagulation for liver fibrosis in patients transplanted for hepatitis C (WAFT-C) trial, and the steroids or pentoxifylline for alcoholic hepatitis (STOPAH) trial. His clinical interests are in the treatment of chronic viral hepatitis infection and in the management of non-alcoholic fatty liver disease. His research interests are focused on the natural history of chronic viral hepatitis, immune responses to viral infection and genetic susceptibility in persistent infections.
Professor Gareth Tudor-Williams is Professor of Paediatric Infectious Diseases at Imperial College London, and Consultant in Paediatric Infectious Diseases at Imperial College Healthcare NHS Trust, St. Mary’s Hospital, London. His interest in paediatric infectious diseases was sparked after having worked for two years in the Himalayas for Save the Children Fund UK. He has spent 30 years contributing to some of the incremental improvements in the management of children living with HIV, and the prevention of vertical transmission from mothers to their infants. He is a Fellow at Duke University, North Carolina and a Visiting Scientist at the National Institutes of Health (NIH), Bethesda, Maryland. During his career, he has worked with the World Health Organisation (WHO) and UNICEF in the Northwest Sindh Province of Pakistan. Since 1994, he has helped run a multi-disciplinary service for children infected with HIV, young people and their families at St. Mary’s Hospital. He was the founding chair of the Children’s HIV Association of the UK and Ireland (CHIVA), alongside being a passionate undergraduate and postgraduate educator, for which he received the IC School of Medicine’s Distinguished Teacher Award in 2017.
Dr Jonathan Wallis is a consultant haematologist. He has a keen interest in transfusion, and his publications include studies on leucodepletion and infection; transfusion-related acute lung injury (TRALI); long term survival after transfusion; and the ‘Tag and Label’ system for blood administration. Dr Wallis is an active member of regional and national transfusion committees, including having chaired the British Blood Transfusion Society (BBTS) Hospital Transfusion Special Interest Group. He also chaired the BBTS Scientific Meetings Administration Committee, and the International Society of Blood Transfusion (ISBT) Working Group on Clinical Transfusion. He initiated and runs the Newcastle course for Transfusion Practitioners and Biomedical Scientists on alternate years, and is also the Associate Editor of Transfusion Medicine.